




Abstract
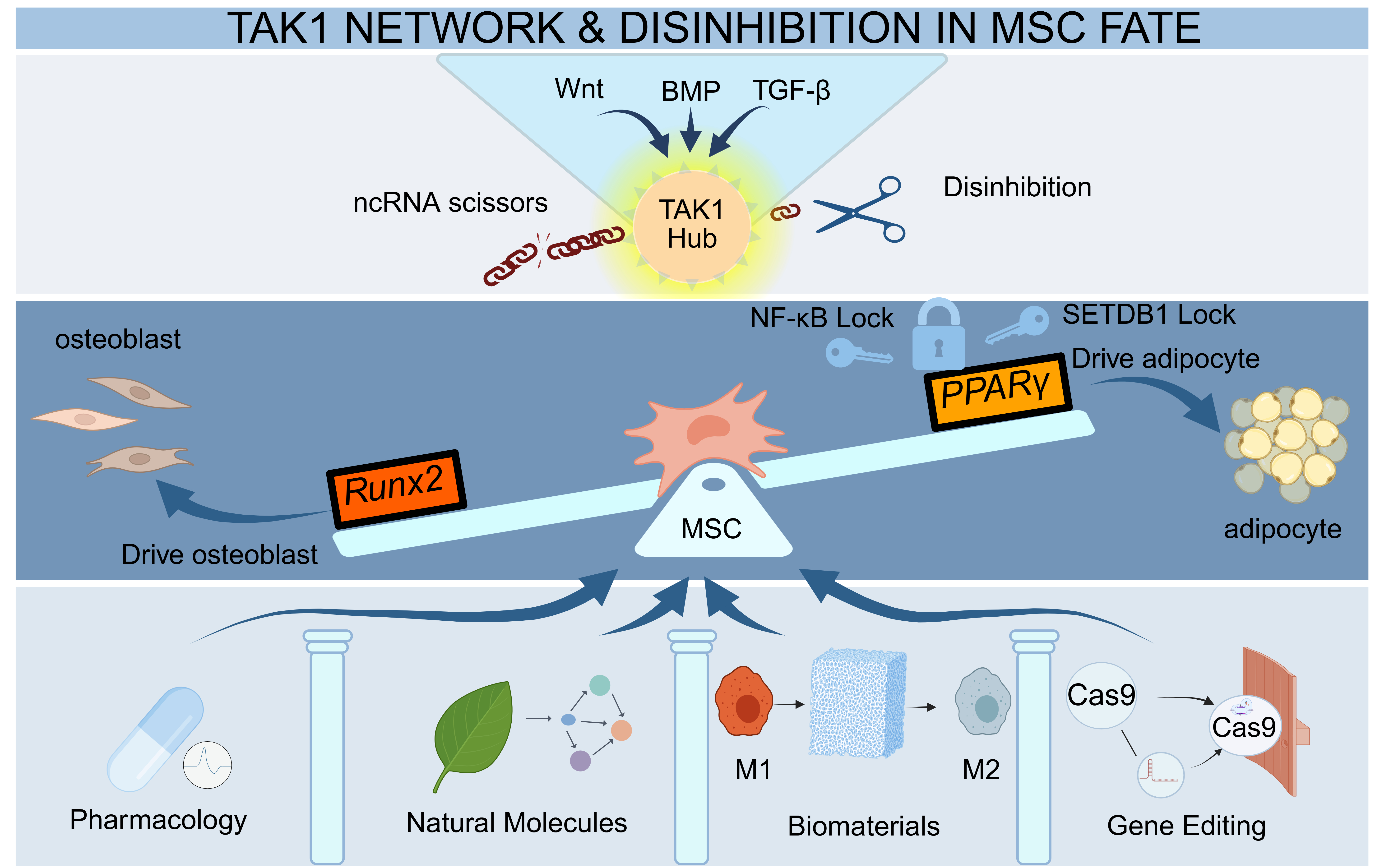
Osteoporosis (OP) represents a systemic failure of bone homeostasis, necessitating a
therapeutic paradigm shift from broad-spectrum anti-resorptive agents to precision
osteo-anabolic strategies. This review analyzes the multidimensional regulatory network
governing osteoblast differentiation, emphasizing the transition from linear pathway
descriptions to systems biology. We first elucidate the hierarchical integration of core
signaling cascades—specifically the "context-dependent" crosstalk among Wnt, BMP, and
TGF-β pathways—and their convergence on the master transcriptional hub, Runx2. A
critical focus is placed on the pathological collapse of the Runx2-PPARγ equilibrium,
which drives the inverse trajectory of osteogenic attenuation and marrow adiposity.
Furthermore, we dissect the post-transcriptional "disinhibition" logic mediated by
non-coding RNAs as a pivotal epigenetic layer. Bridging mechanistic insights with
clinical translation, we classify emerging targeted strategies into three precision
dimensions: pathology-dependent pharmacological reprogramming, osteoimmunomodulatory
biomaterials with spatiotemporal responsiveness, and "scarless" gene editing
technologies. Finally, we critically address pivotal translational
bottlenecks—specifically the delivery barriers across the mineralized matrix and
off-target safety risks—proposing that future therapeutics must evolve toward the
precision homeostatic remodeling of the skeletal microenvironment.
Keywords:OP; osteoblast; differentiation; signaling; therapeutic paradigm
Introduction
The skeletal system transcends its role as a mere physical framework for mechanical support to function as a highly active metabolic organ. Its physiological homeostasis hinges on the delicate dynamic equilibrium of bone remodeling—specifically, the tight coupling between osteoblast-mediated bone formation and osteoclast-mediated bone resorption. The uncoupling of this balance, driven by aging or hormonal fluctuations, precipitates OP, a condition characterized by systemic bone loss [1,2] . Clinically defined by reduced bone mass, microarchitectural deterioration, and a significantly elevated risk of fragility fractures, OP presents persistent therapeutic challenges. Current pharmacotherapies, such as bisphosphonates, are hampered by a lack of cellular specificity, leading to severe complications including osteonecrosis of the jaw [2] . To overcome these limitations, the therapeutic paradigm must shift from broad phenotypic inhibition to the precise dissection of deep molecular networks, thereby facilitating the development of novel osteo-anabolic strategies.
Emerging evidence highlights the profound "context-dependency" of osteogenic regulation. The dualistic "double-edged sword" nature of TGF-β—balancing homeostasis against pathological induction [3] —and the strict reliance of Wnt5a signaling flow on specific receptor combinations [4] underscore the inadequacy of traditional linear intervention strategies. Furthermore, extensive pathway crosstalk and endogenous feedback loops establish a highly intricate regulatory topology [5] .
Consequently, this review systematically deconstructs the multidimensional network of osteogenic differentiation across three critical dimensions: core signaling integration, transcriptional hubs, and non-coding RNA regulation. Our objective is to provide a theoretical foundation for the next generation of precision bone regenerative therapies.
Pathogenesis of OP
The core of OP lies in the disruption of the balance between bone formation and bone resorption. This is not only due to the disorder of osteoblast differentiation, function and progenitor cell pool, but also involves the vicious cycle such as the dysregulation of osteogenic and osteoclasts coupling and adipogenesis.
Overview of Osteoblast Differentiation and Osteogenic Function
Osteoblasts, the pivotal effectors of bone formation, originate from multipotent MSCs [6] . The differentiation of osteoblasts is a highly orchestrated, multi-stage cascade. Initiated by signaling pathways such as Bone Morphogenetic Protein (BMP) and Wnt, MSCs commit to the osteoprogenitor lineage and proliferate into pre-osteoblasts. Under the command of master transcriptional regulators Runx2 and Osterix, these cells ultimately mature into highly secretory osteoblasts [7] . Following completion of bone matrix synthesis, a subset of osteoblasts becomes embedded within the mineralized matrix and differentiates into osteocytes, which constitute the majority of bone cells. These osteocytes function as primary mechanosensors and central coordinators of signaling networks within bone tissue. The remaining osteoblasts are either eliminated through apoptosis or undergo phenotypic conversion into quiescent bone lining cells that cover the bone surface [8] .
Functionally, osteoblasts are primarily responsible for synthesizing the organic matrix—predominantly Type I collagen—to construct the osteoid scaffold, while concurrently secreting non-collagenous proteins, such as osteocalcin and osteopontin, to modulate the local microenvironment. Subsequently, they drive mineralization by releasing calcium- and phosphate-rich matrix vesicles and expressing phosphatases, notably alkaline phosphatase (ALP). These mechanisms catalyze the ordered deposition of hydroxyapatite crystals within collagen fibrils, thereby transforming soft osteoid into rigid osseous tissue [9] .
Dysregulation of Osteoblast-Osteoclast Homeostasis and Mechanisms of Bone Loss
The pathological essence of OP lies in the dismantling of the "osteoblast-osteoclast" coupling mechanism and a deviation in the "osteogenic-versus-adipogenic" differentiation trajectory. Collectively, these factors establish a catabolic dominance within the bone microenvironment.
The decline in osteogenic capacity stems not merely from compromised terminal differentiation efficiency, but more fundamentally from the exhaustion of the upstream progenitor reservoir. The canonical Wnt ligand, Wnt10b, has been identified as a pivotal regulator maintaining the homeostasis of adult bone marrow mesenchymal progenitor cells (MPCs). Deficiency in Wnt10b confers an instructive "biphasic phenotype" in mice: an initial, transient increase in bone mass during juvenility driven by accelerated differentiation, followed by accelerated physiological bone decay as the progenitor pool is prematurely depleted, failing to replenish functional osteoblasts [1] . This quantitative depletion is frequently accompanied by qualitative deterioration—specifically, a pathological drift in the MSC differentiation trajectory. As the antagonistic equilibrium between Runx2 and the adipogenic factor PPARγ collapses, ectopic accumulation of bone marrow adipocytes occurs. Far from being metabolically inert bystanders, these adipocytes actively secrete RANKL to drive osteoclastogenesis, establishing a vicious feedback loop of "lipid infiltration aggravating bone resorption" [10] .
Communication mismatch between osteoblasts and osteoclasts represents another dimensional mechanism underlying bone loss. Beyond the classical imbalance of the RANKL/OPG axis, non-canonical Wnt signaling exerts a more cryptic yet critical influence. Osteoblast-lineage-derived Wnt5a acts via a paracrine mechanism to engage the Ror2 receptor on osteoclast precursors, upregulating RANK transcription, thereby significantly amplifying osteoclastic activity [11] . This stands in sharp contrast to Wnt5a's osteogenic role under specific receptor contexts, suggesting that the Wnt5a-Ror2 axis may function as a pathological "accelerator" of bone resorption. Such signaling exhibits high context-dependency: within the multiple myeloma (MM) microenvironment, tumor cells inversely suppress Ror2 expression in osteoprogenitors to blockade non-canonical Wnt signaling, thereby exacerbating osteogenic suppression within osteolytic lesions [12] . These diametrically opposed molecular consequences profoundly underscore the functional plasticity of signaling pathways across distinct microenvironmental configurations.
Key Signaling Pathways Governing Osteoblast Differentiation
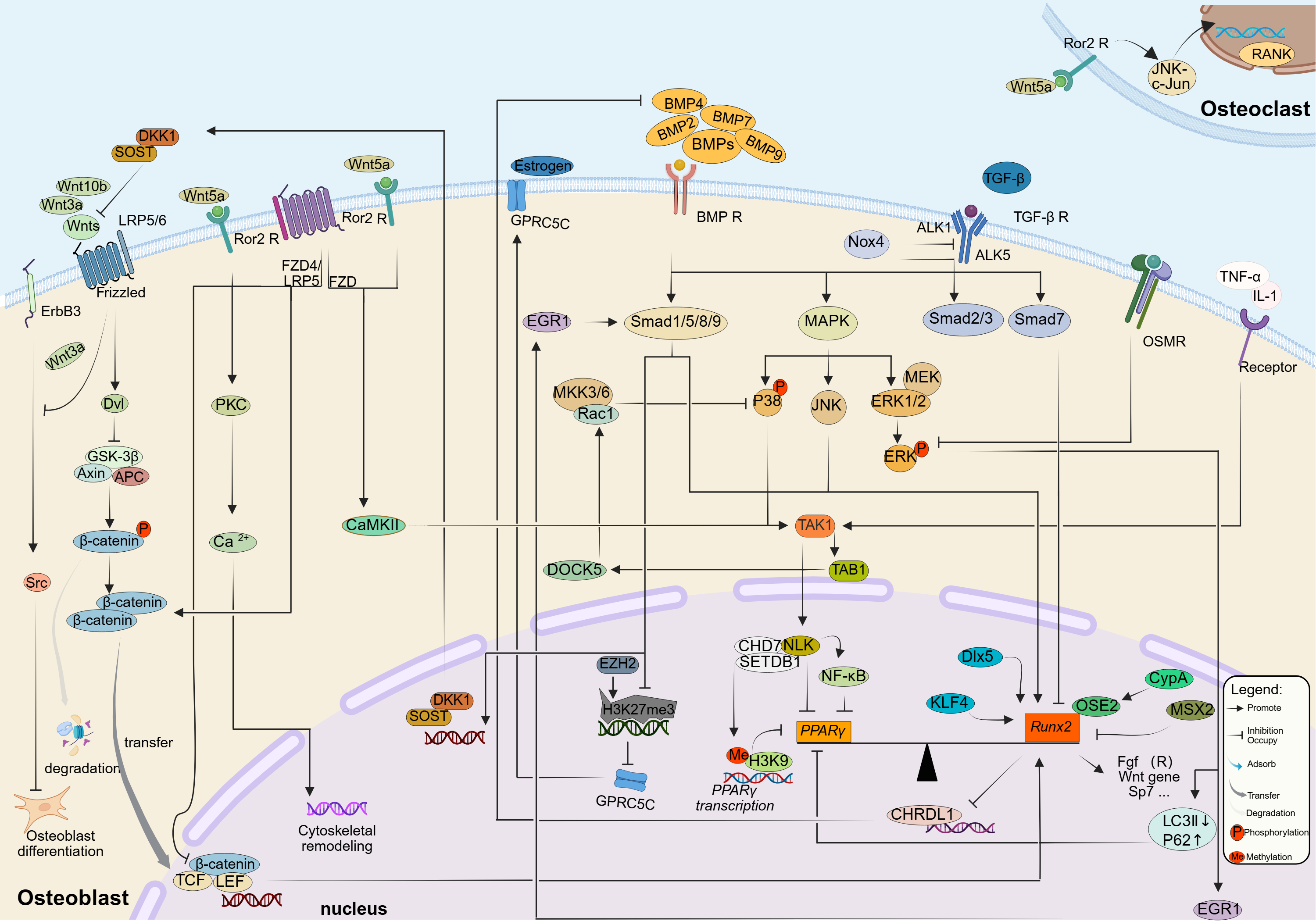
A schematic overview of the interconnected Wnt, BMP/TGF-α, and downstream transcriptional regulatory modules governing osteoblast differentiation is presented in Figure 1.
Figure 1. The multidimensional signaling topology and hierarchical
integration of the osteoblast differentiation network.
This schematic illustrates the systems-level logic governing osteogenic
fate, highlighting the transition from extracellular signal integration to
nuclear transcriptional reprogramming and epigenetic locking. (A) Membrane
Reception and Context-Dependent Crosstalk: The network initiates with the
spatiotemporal integration of core signals. Canonical Wnt signaling (left)
is triggered by ligands such as Wnt10b and Wnt3a, which stabilize β-catenin
to drive progenitor maintenance. In contrast, non-canonical Wnt5a exhibits
profound "context-dependency": it engages the Ror2/FZD receptor complex to
activate the osteogenic PKC and CaMKII axes, while acting via a paracrine
mechanism on osteoclast precursors (top right) to upregulate RANK and
amplify resorption. Meanwhile, the BMP and TGF-β pathways (in the middle)
not only activate their respective pathways but also converge at the MAPK
effector factor, among which the BMP2-activated pathway is strictly
regulated by auxiliary sensors (such as GPRC5C), which is epigenetically
derepressed upon the removal of EZH2-mediated H3K27me3 marks. (B)
Cytoplasmic Convergence Hubs: Distinct signals converge on TAK1, a pivotal
kinase nexus that integrates inputs from Wnt (via CaMKII), BMP/TGF-β, and
inflammatory cytokines (TNF-α/IL-1) to prioritize cell survival and
differentiation. Signal flux is further fine-tuned by intracellular
feedback loops: EGR1 acts as a "cytosolic adapter" bridging ERK to Smad
activation (positive feedback), whereas OSMR functions as a metabolic
brake by suppressing ERK phosphorylation and blocking autophagic flux
(indicated by LC3II reduction and P62 accumulation). (C) The Runx2-PPARγ
Equilibrium and Epigenetic Blockade: In the nucleus, Runx2 serves as the
master transcriptional hub, synergized by co-factors and self-reinforced
by repressing the antagonist CHRDL1. The ultimate lineage commitment
hinges on the reciprocal equilibrium between Runx2 and the adipogenic
regulator PPARγ. The diagram highlights a critical pathological
mechanism: the collapse of this balance is prevented by a Wnt5a-induced
epigenetic blockade, where the NLK-CHD7-SETDB1 complex catalyzes H3K9
methylation at the PPARγ promoter, thereby silencing adipogenesis to
consolidate the osteogenic program.
Wnt Signaling Pathway: Crosstalk between Canonical and Non-canonical Cascades
Wnt signaling functions not as a simple linear directive, but as a dynamic logic matrix governing cell fate. The canonical pathway operates primarily by disrupting the homeostatic degradation of β-catenin. Upon binding of Wnt ligands to the Frizzled receptors and LRP5/6 co-receptors, the signal is transduced via the key adaptor protein Dishevelled (Dvl). This disrupts the functionality of the Axin/GSK-3β/APC destruction complex, allowing β-catenin to accumulate in the cytoplasm, translocate to the nucleus, and associate with TCF/LEF transcription factors to initiate target gene expression [13~15] . Conversely, the non-canonical pathway, with Ror2 acting as a central hub, circumvents β-catenin reliance, instead directly orchestrating cytoskeletal remodeling via PKC activation and calcium flux [16,17] . Although these pathways generally function independently, they often converge through deep signal crosstalk within the multidimensional regulatory networks of cell differentiation to dictate the final cellular response.
Distinct ligands have evolved highly specific regulatory strategies centered on the "β-catenin stability" node. Wnt3a not only directly activates canonical signaling but also alleviates the repression of osteogenesis by downregulating the ErbB3/Src axis [18] . However, as this finding relies predominantly on ALP activity—an early differentiation marker—its actual contribution to terminal events, such as mineralized nodule formation, necessitates multidimensional validation. Meanwhile, Wnt10b has been established as a pivotal factor maintaining the MPC pool via the canonical pathway; its deficiency precipitates progenitor exhaustion and age-related bone loss [1,19] . While the molecular nuances of Wnt10b-driven self-renewal await complete dissection via in vivo lineage tracing, evidence suggests that Wnt10b-induced, VEGF-mediated angiogenesis may serve a mechanistic role beyond mere blood supply, potentially coupling vascular niche signals to stem cell maintenance [20] .
The functional paradigm of the non-canonical ligand Wnt5a profoundly illustrates the principle of signaling "context-dependence." The qualitative nature of Wnt5a signaling is contingent upon the cell surface receptor profile: when bound to the receptor tyrosine kinase-like orphan receptor 2 (Ror2), it inhibits TCF-mediated transcription following β-catenin stabilization; conversely, in cells co-expressing FZD4 and LRP5, it switches roles to become a canonical activator [4] . During MSC osteogenic differentiation, Wnt5a activates the CaMKII-TAK1-NLK axis via Ror2/FZD, thereby suppressing adipogenesis and promoting osteogenesis [21] . While this mechanism has been elucidated in cell lines, the suppression of PPARγ poses a risk of systemic metabolic perturbation, highlighting the need for tissue-specific targeting strategies in future translational research. This signaling plasticity is further evident in MM, where tumor cells blockade osteogenesis by suppressing Ror2 expression in osteoprogenitors; restoring this pathway can reverse the inhibition [12] . However, given the in vitro basis of these findings, their validity and therapeutic potential within the complex in vivo bone marrow microenvironment remain to be verified in animal models.
In vivo studies further underscore the dual signaling activity of Wnt5a. Systemic overexpression leads to skeletal deformities in mice yet activates β-catenin reporters in the cranial dura mater [22] . This suggests that Wnt5a can convert into a canonical agonist under specific conditions, although whether this causes delayed cranial ossification requires confirmation via conditional knockout models. Developmentally, Wnt5a and Wnt5b coordinate the rhythm of chondrocyte proliferation and differentiation through the differential and antagonistic regulation of Sox9 and the cell cycle [23] . Furthermore, the effects of Wnt5a are not confined to the osteogenic lineage; osteoblast-derived Wnt5a can be captured via paracrine signaling by Ror2 on osteoclast precursors, activating the JNK-c-Jun pathway and upregulating RANK expression [11] . This interaction between osteoblasts and osteoclasts significantly amplifies the complexity of therapeutic targeting.
While the Wnt network constitutes the cornerstone of bone remodeling regulation, it does not operate in a hermetic vacuum. To achieve the precise spatiotemporal coupling of bone metabolism, Wnt signaling must undergo deep synergy and integration with other critical signaling clusters, such as the BMP/TGF-β superfamily.
BMP/TGF-β Signaling Pathways
As central pillars of the TGF-β superfamily, the Transforming Growth Factor-β (TGF-β) and BMP signaling pathways orchestrate MSC osteogenesis and differentiation. They operate through a dualistic mechanism involving the canonical Smad-dependent axes (TGF-β/Smad2/3 and BMP/Smad1/5/8) and non-canonical cascades, such as p38 MAPK and JNK, playing distinct yet critical roles [5,24] .
BMP signaling functions as a potent osteo-inductive force through the synergy of these pathways. Evidence suggests a temporal dichotomy in regulation: p38 preferentially modulates early osteogenic markers, whereas JNK governs the expression of late-stage markers [25] . This intricate spatiotemporal specificity underscores the primary challenge in clinical translation: biological efficacy is strictly governed by local concentration and signaling kinetics. Physiological BMP signaling operates with precise spatiotemporal confinement, a pattern that traditional high-dose bolus administration fails to recapitulate, frequently resulting in adverse effects such as ectopic ossification [26] . To circumvent this delivery dilemma, intervention at endogenous regulatory nodes within the BMP pathway has emerged as a critical strategy. The output intensity of BMP signaling is constrained by a sophisticated endogenous network; for instance, DOCK5 has been identified as a key negative checkpoint for BMP2 signaling. It selectively attenuates p38 activity via the inhibition of the Rac1-MKK3/6 axis, while notably sparing the canonical Smad1/5/9 phosphorylation [27] . The delineation of such negative feedback mechanisms suggests that targeting endogenous inhibitory nodes could sensitize cells to BMP signals, offering a novel paradigm to enhance safety by reducing the requisite therapeutic dosage of exogenous BMP.
In contrast to the unequivocal osteogenic potency of BMP, TGF-β exerts a quintessential "double-edged sword" effect on bone metabolism, with terminal outcomes highly contingent upon cell type and microenvironmental context [3,5] . For instance, TGF-β1 suppresses osteogenic differentiation in dental follicle cells [28] , whereas the blockade of the TGF-β Type I receptor ALK5 downregulates the osteogenic capacity of human MSCs [29] , implying that a basal level of TGF-β signaling is indispensable for osteogenesis. These seemingly contradictory findings reflect the profound lineage specificity of the pathway. Consequently, single-dimensional observations are often prone to bias; resolving these discrepancies necessitates the establishment of a unified validation framework utilizing rigorous Gain- and Loss-of-function assays. This complexity is further highlighted by Nox4-associated paradoxes: while Nox4 deficiency leads to TGF-β receptor downregulation, it is counterintuitively accompanied by elevated Smad2 phosphorylation [30] . Such contradictions likely stem from the differential engagement of distinct receptor subtypes, such as the balance between ALK5 and ALK1 activation [3] .
Integration of Wnt, BMP, and TGF-β Signaling Networks
The Wnt, BMP, and TGF-β signaling pathways do not operate in isolation; rather, they coalesce into a highly dynamic and "context-dependent" regulatory network facilitated by receptor sharing, nodal convergence, and transcriptional integration. This networked interactivity constitutes the central logic governing the precise orchestration of bone formation and homeostasis [2,5] .
A pivotal locus for this multi-pathway integration is the shared intracellular signaling hub, most notably Transforming Growth Factor-β Activated Kinase 1 (TAK1). TAK1 functions not only as the requisite upstream kinase for the non-canonical BMP/TGF-β pathways [24] but also as the nexus integrating Wnt5a signaling (via the CaMKII-TAK1-NLK axis) with inflammatory cascades (TNF-α/IL-1) [21,27] . Consequently, TAK1 acts as a molecular crossroad, synthesizing inputs from Wnt, BMP/TGF-β, and inflammatory stimuli to dictate decisive cell fate outcomes.
Inter-pathway crosstalk initiates at the level of ligand expression and activity modulation. BMP signaling is known to upregulate the Wnt antagonists SOST and DKK1, a negative feedback mechanism designed to prevent aberrant bone overgrowth [5,24] . Conversely, Runx2 transcriptionally represses the BMP antagonist CHRDL1, establishing a Runx2/CHRDL1/BMP4 positive feedback loop [31] . Within the nucleus, Runx2 serves as the master transcriptional nexus: it is a direct target of BMP/Smad1/5/8, is synergistically promoted by Wnt/β-catenin, and is negatively regulated by TGF-β-induced Smad7 [28] . This intricate integration explains why a single molecular entity can elicit diametrically opposed biological outputs depending on the specific signaling milieu.
Existing research paradigms, largely constrained by linear analysis of single pathways, are increasingly insufficient to address this biological reality. Future breakthroughs necessitate the adoption of systems biology approaches to elucidate the mechanism of signal "priority sorting" at critical nodes like TAK1 under pathological conditions. Such insights will define the theoretical foundation for the next generation of precision therapies for skeletal diseases [2,3] .
Transcriptional Regulation and Key Factors in Osteogenesis
Runx2 as the Master Regulator of Osteoblast Differentiation
Runt-related transcription factor 2 (Runx2) serves as a quintessential "master regulator" of MSC osteogenic lineage commitment. Its orchestration spans the entire continuum of cell proliferation, matrix synthesis, and signaling remodeling, exerting precise spatiotemporal control over the osteogenic program [28,31~33] .
During early differentiation, Runx2 promotes progenitor proliferation by modulating FGF receptors and Wnt genes, subsequently inducing Sp7 (Osterix) expression to drive maturation. Remarkably, during endochondral ossification, it is capable of inducing the transdifferentiation of hypertrophic chondrocytes into osteoblasts [32] . However, this potency exhibits a distinct tissue-specific dichotomy: while Runx2 activity protects against OP in the osteogenic lineage, its aberrant expression in articular cartilage acts as a pathogenic driver of osteoarthritis.
To consolidate differentiation signals, Runx2 actively engineers positive feedback loops to remodel the microenvironment. Research has uncovered a "Runx2/CHRDL1/BMP4" axis, wherein Runx2 transcriptionally represses the BMP antagonist CHRDL1, thereby unleashing BMP4/Smad signaling [31] . However, chromatin immunoprecipitation (ChIP) assays have demonstrated relatively low enrichment of Runx2 at the CHRDL1 promoter, failing to conclusively verify direct binding. This suggests that the observed repression may be indirect; future investigations should incorporate high-resolution techniques, such as Chromatin Conformation Capture (Hi-C), to elucidate the physical interaction mechanisms underlying this regulation.
Beyond its expression levels, the transcriptional efficacy of Runx2 is contingent upon the dynamic assembly of transcriptional complexes. Kruppel-like factor 4 (KLF4) has been identified to co-occupy over 80% of osteogenic enhancer regions with Runx2, synergistically driving downstream gene expression [34] . Similarly, Cyclophilin A (CypA) significantly potentiates the DNA-binding affinity of Runx2 to OSE2 elements via direct protein interaction [35] . A critical knowledge gap with high translational relevance remains in this field: it is currently undefined whether the peptidyl-prolyl cis-trans isomerase (PPIase) activity of CypA is substantively involved in the conformational regulation of Runx2. Resolving this ambiguity is pivotal for drug discovery, as it will determine whether small-molecule design should target the enzymatic active site of CypA or its protein-protein interaction interface with Runx2.
On the counter-regulatory side of the network, Runx2 is subject to rigorous competitive constraints to maintain homeostasis. During development, the homeobox proteins Dlx5 and Msx2 exert activating and repressive functions, respectively, through competitive binding to the Runx2 promoter [36] . In pathological contexts, TGF-β-induced Smad7 functions as a potent molecular "brake," blockading Runx2-mediated activation of downstream osteogenic genes [28] .
Other Regulatory Factors
Although Runx2 constitutes the central nexus of osteogenic differentiation, the ultimate determination of osteogenic fate within complex pathophysiological milieus hinges on the "fine-tuning" and "remodeling" of core signals by auxiliary factors.
To surmount the activation threshold required for differentiation, cells exploit cross-pathway crosstalk to achieve cascade amplification. Early Growth Response 1 (EGR1) has been identified not merely as a transcription factor, but as a pivotal "cytosolic adapter" bridging the non-canonical and canonical BMP pathways. Research demonstrates that the potent osteo-inductive factor BMP9 rapidly induces EGR1 expression via the MEK/ERK axis. Newly synthesized EGR1 physically associates with SMAD1/5 in the cytoplasm, maintaining SMAD activity by sterically hindering phosphatase-mediated dephosphorylation, thereby establishing an "ERK-EGR1-SMAD" positive feedback loop [37] . While this mechanism elegantly elucidates the source of BMP9 signaling persistence, it leaves a significant knowledge gap: the specific identity of the phosphatase competitively inhibited by EGR1 remains unknown.
Beyond signal amplification, osteoblasts require the capacity to interrogate the microenvironment and epigenetic landscape; the orphan receptor GPRC5C functions as this "environmental sensor." In the quiescent state, GPRC5C expression is stringently repressed by EZH2-mediated H3K27me3 modifications. Upon the alleviation of this repression by BMP2, GPRC5C rapidly drives matrix mineralization by responding to estrogen signaling [38] . Currently, the "orphan receptor" status of GPRC5C constitutes a formidable bottleneck for clinical translation. Research must shift from phenotypic description to mechanistic dissection, utilizing high-throughput CRISPR screening or ligand-fishing strategies to identify endogenous ligands and downstream G-protein coupling. This will delineate specific signal transduction pathways, laying the foundation for the development of agonists that mimic osteo-anabolic metabolism.
On the negative regulatory spectrum, the Oncostatin M Receptor (OSMR) functions as a critical "brake," underscoring the intricacy of the osteogenic network through the inverse utilization of shared signaling nodes. Diametrically opposed to the reliance of EGR1 on ERK phosphorylation for activation, aberrantly elevated OSMR in OP models suppresses autophagic flux (manifested by reduced LC3II and accumulated P62) by downregulating p-ERK levels, thereby blocking osteogenesis and promoting adipogenesis [39] . This profoundly reveals the duality and context-dependence of ERK signaling: it serves as the requisite "engine" for EGR1-mediated amplification yet acts as a key node "extinguished" by OSMR to arrest differentiation. Although targeting OSMR silencing has achieved an "osteo-anabolic and anti-adipogenic" effect in ovariectomized (OVX) models, this contradicts earlier findings of impaired bone formation in systemic knockout mice. This discrepancy strongly suggests a compensatory interplay between the local microenvironment and systemic regulation.
In synthesis, the cytosolic signal maintenance by EGR1, the epigenetic derepression of GPRC5C, and the metabolic autophagy regulation by OSMR collectively construct a multidimensional auxiliary regulatory matrix beyond the core Runx2 axis. This serves as a caveat that therapeutic strategies for skeletal diseases must transcend the linear reductionism of single-target approaches and shift towards the systemic remodeling of network topology.
The Runx2-PPARγ Axis: A Critical Switch between Osteogenesis and Adipogenesis
The lineage commitment of bone marrow MSCs adheres to a zero-sum logic of mutual exclusivity, a balance primarily maintained by the reciprocal inhibition between the master osteogenic transcription factor, Runx2, and the core adipogenic regulator, PPARγ. In the context of OP and skeletal aging, the collapse of this equilibrium—characterized by the arrest of the Runx2-driven osteogenic program and the concurrent onset of PPARγ-mediated marrow adiposity—constitutes the fundamental pathological basis of bone loss.
Regulation of this equilibrium by canonical Wnt signaling exhibits complex "bidirectional effects." Although Wnt10b theoretically consolidates osteogenic dominance by suppressing PPARγ and its downstream adipogenic targets [20] , in vivo and in vitro experiments point to a significant mechanistic dichotomy. While Wnt10b deficiency precipitates severe bone loss in mice, the corresponding bone marrow stromal cells paradoxically exhibit diminished adipogenic potential in culture [1] . This phenomenon suggests that the core in vivo function of Wnt10b may not be merely to drive differentiation polarity, but to maintain the quantity and vitality of the undifferentiated progenitor pool. Consequently, the exhaustion of the MSC reservoir following Wnt10b loss yields consequences far more severe than a simple shift in differentiation bias. Future research must therefore transcend the limitations of single-marker detection, employing lineage tracing technologies to precisely delineate the temporal switching mechanism by which Wnt signaling governs the transition between "stemness maintenance" and "differentiation initiation."
In contrast to the direct transcriptional regulation by the canonical pathway, non-canonical Wnt signaling imposes "gene silencing" on PPARγ through sophisticated epigenetic mechanisms. Wnt5a-activated NLK kinase functions as a nuclear molecular scaffold, recruiting the histone methyltransferase SETDB1 and the chromatin remodeler CHD7 to the PPARγ promoter. This complex catalyzes H3K9 methylation, leading to chromatin compaction and transcriptional silencing [21] . This "epigenetic blockade" arrests the adipogenic program at its source. Given that PPARγ is also a systemic hub for insulin sensitivity, the breakthrough for clinical translation lies in developing interfering molecules that specifically deconstruct the "NLK-SETDB1-PPARγ" ternary complex. The objective is to achieve a safe "functional decoupling" that preserves osteogenic benefits while mitigating systemic metabolic risks, rather than blindly ablating the entire PPARγ pathway.
Acting as a terminal checkpoint linking microenvironmental stress to cell fate, metabolic autophagic flux plays an equally decisive role. Pathologically elevated OSMR blocks autophagic flux by inhibiting ERK activity, thereby preventing the clearance of intracellular lipid droplets via "lipophagy." The resulting accumulation of lipotoxicity exerts a suppressive effect on Runx2 activity, inversely solidifying the dominance of PPARγ [39] .
In summary, the competition between Runx2 and PPARγ is substantively a systemic confrontation spanning transcriptional, epigenetic, and metabolic dimensions. Future therapeutic strategies must evolve from a simplistic view of "reciprocal trade-offs" between single factors toward a holistic remodeling of the entire regulatory chain: "Progenitor Pool – Epigenetic Blockade – Autophagic Flux."
Non-coding RNA-mediated Post-transcriptional Regulatory Networks
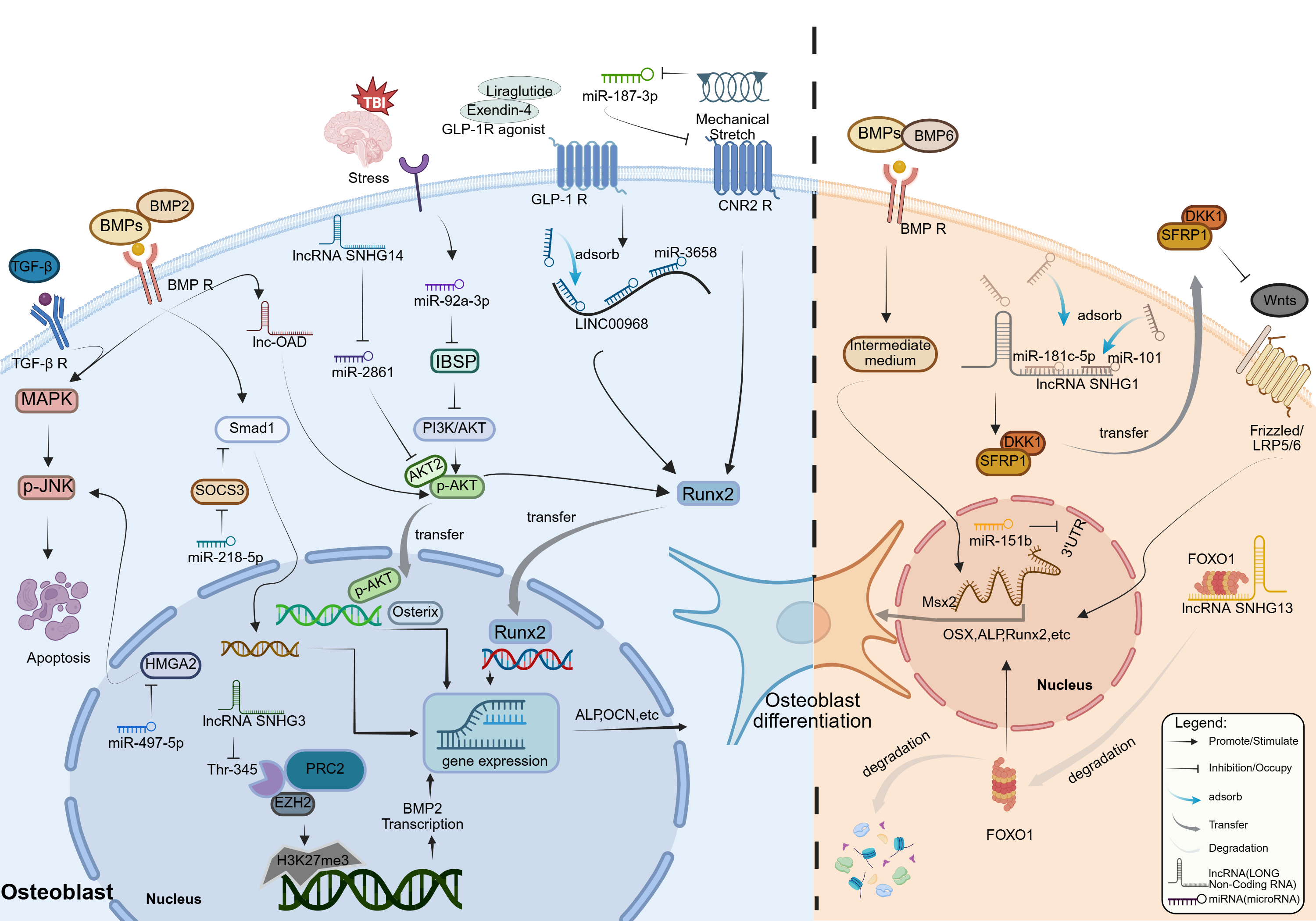
A schematic overview of the miRNA/lncRNA-centered post-transcriptional regulatory network and its major signaling/transcriptional nodes in osteoblast differentiation is presented in Figure 2.
Figure 2. The post-transcriptional logic and epigenetic
architecture of non-coding RNA-mediated osteogenic fate determination.
This schematic delineates the dichotomous regulatory landscape governed by
non-coding RNAs, contrasting the logic of "disinhibition" (Left, Blue
panel) against "suppression" (Right, Orange panel) in the control of
osteoblast differentiation. (A) The Osteo-Anabolic Network: Disinhibition
and Epigenetic Derepression. The left panel illustrates how osteogenic
signals are safeguarded through the neutralization of endogenous
inhibitors. Signaling Disinhibition: MicroRNAs act as checkpoints to
release signaling blockades. miR-218-5p targets the inhibitor SOCS3,
thereby unleashing Smad1 activity; miR-92a-3p, triggered by TBI stress,
suppresses IBSP to reactivate the PI3K/AKT axis; and mechanical stretch
downregulates miR-187-3p, relieving the repression on the CNR2 receptor.
ceRNA & Epigenetic Scaffolds: Long non-coding RNAs (lncRNAs) function
through different structural mechanisms. In the cytoplasm, SNHG14 and
LINC00968 act as competitive endogenous RNAs to protect the transcripts
of AKT2 and Runx2 by inhibiting miR-2861 and adsorbing miR-3658,
respectively. In the nucleus, SNHG3 acts as an epigenetic decoy, anchoring
EZH2 (of the PRC2 complex) to prevent H3K27me3 deposition at the BMP2
promoter, thus maintaining transcriptional accessibility. (B) The
Osteo-Suppressive Network: Feedback Paradox and Degradation. The right
panel depicts mechanisms leading to differentiation arrest. The ceRNA
Paradox: Highlighting functional divergence, SNHG1 adsorbs miRNAs such as
miR-181c-5p and miR-101, preventing the inhibition of Wnt pathway
antagonists DKK1 and SFRP1 (thereby stabilizing them), and ultimately
leading to the blocking of the classical Wnt signaling pathway. Targeted
Degradation: SNHG13 operates via a nucleic-acid-independent mechanism,
directly binding FOXO1 to induce its ubiquitin-proteasomal degradation.
Concurrently, miR-151b clamps the 3'UTR of Msx2, severing the downstream
transcriptional cascade essential for mineralization.
miRNA-mediated Targeting of Signaling Nodes and Transcription Factors
MicroRNAs (miRNAs) function as highly efficient molecular switches that safeguard osteogenic signal flux through the mechanism of "disinhibition"—the precise clearance of endogenous inhibitors within signaling networks. Research indicates that miRNAs specifically target the molecular "brakes" of signaling pathways, thereby alleviating blockades on core cascades including BMP, MAPK, and PI3K/AKT. Specifically, miR-218-5p directly binds and suppresses the Suppressor of Cytokine Signaling 3 (SOCS3). This interaction not only releases the inhibition of SMAD1 downstream of BMP signaling but also disrupts the negative feedback loop promoting osteoclastogenesis, effectively remodeling metabolic balance in postmenopausal OP (PMOP) models [40] . Similarly, at the level of kinase regulation, miR-497-5p targets High Mobility Group AT-Hook 2 (HMGA2) to significantly attenuate JNK pathway phosphorylation, inhibiting apoptosis and promoting differentiation [41] . In the context of Traumatic Brain Injury (TBI) stress, miR-92a-3p activates repressed PI3K/AKT signaling by inhibiting Integrin Binding Sialoprotein (IBSP), thereby accelerating osteogenesis [42] .
While existing studies have outlined this regulatory framework, their explanatory power remains constrained by the limitations of a reductionist perspective. The preponderance of evidence is derived from isolated precursor cell models [41,42] , which artificially strip away the essential paracrine interactions with osteoclasts and vascular endothelial cells inherent to the bone microenvironment. More paradoxically, the inverse expression trends of SOCS3—high in bone tissue versus low in peripheral blood [40] —strongly suggest a mechanistic decoupling between local microenvironments and systemic regulation. Consequently, future mechanistic dissection must transcend single-dimensional expression screening. By leveraging spatial transcriptomics to map in situ interaction landscapes under pathological conditions, combined with multicellular co-culture systems, research can define the authentic efficacy of miRNAs while respecting the complexity of tissue communication.
Beyond modulating upstream signal flux, miRNAs directly dictate the abundance of transcription factors and membrane receptors, serving as conduits linking extracellular physical fields to intracellular genetic programs. At the transcriptional terminus, miR-151b has been identified as a potent repressor of osteogenic differentiation. Its aberrant upregulation in osteoporotic environments cascades to shut down the BMP-6-induced OSX/ALP/Runx2 expression program by specifically clamping the 3'UTR of the homeobox transcription factor Msx2 [43] . At the membrane receptor interface, miRNAs mediate rapid mechanotransductive responses: mechanical stretching alleviates the blockade on Cannabinoid Receptor 2 (CNR2) by downregulating miR-187-3p, subsequently activating the "Stress-CNR2-Runx2" axis to drive bone formation [44] .
Although these findings have pinpointed critical targets, current mechanistic resolutions remain incomplete. It remains inconclusive whether specific MAPK isoforms are involved in the downstream cascade of Msx2 [43] . Furthermore, studies on CNR2 have simulated only tensile stress, neglecting the physiological compressive forces primarily borne by the skeleton [44] . In light of this, subsequent work must delineate the kinase interaction networks downstream of Msx2 and incorporate 3D bioreactor models capable of simulating complex loading environments—integrating composite tension-compression and fluid shear stress—to comprehensively evaluate the therapeutic potential of these miRNA targets under authentic physiological mechanical conditions.
Roles of lncRNAs in Determining Osteogenic Fate
In contrast to microRNAs, which predominantly fine-tune gene expression at the post-transcriptional level, long non-coding RNAs (lncRNAs) leverage their complex secondary structures and diverse subcellular compartmentalization to orchestrate a multidimensional regulatory architecture spanning from epigenetics to protein modification. By functioning as molecular sponges, scaffolds, and guides, lncRNAs establish a sophisticated layer of control over cellular fate.
As the canonical paradigm of lncRNA-mediated osteogenesis, the competing endogenous RNA (ceRNA) mechanism operates by competitively sequestering miRNAs to alleviate the repression of downstream osteogenic mRNAs. In osteogenic regeneration models, LINC00968 exhibits distinct positive regulatory functionality: under GLP-1R agonist induction, it specifically absorbs miR-3658, releasing the repressed core factor Runx2 and thereby driving the mineralization and regeneration of human dental pulp stem cells (hDPSCs) [45] . However, functional divergence within the SNHG family reveals the high nonlinearity of this network. While SNHG14 activates the AKT2 pathway by blocking miR-2861, SNHG1 conversely releases Wnt inhibitors such as SFRP1 and DKK1 by sequestering miR-181c-5p or miR-101, leading to unexpected osteogenic arrest [46] . This intra-familial functional dichotomy indicates that ceRNA efficacy is not dictated solely by the expression fluctuations of a single molecule, but is strictly governed by the rigorous stoichiometry between the lncRNA, the miRNA, and their targets.
Beyond RNA-RNA interactions, lncRNAs exert control through independent dimensions by physically associating with proteins to modulate epigenetic modifications and signal transduction. In the nucleus, lncRNAs can serve as molecular decoys for epigenetic complexes. SNHG3 has been confirmed to directly anchor EZH2, the catalytic subunit of the PRC2 complex. By occupying the Thr-345 phosphorylation site, SNHG3 imposes steric hindrance that blocks EZH2-mediated H3K27me3 repressive methylation at the BMP2 promoter region [47] . The dissolution of this "SNHG3-EZH2-H3K27me3" axis directly reactivates the transcriptional competency of BMP2-Smad1/5 signaling. Conversely, cytosolic lncRNAs function more as signaling rheostats. lnc-OAD has been identified as an essential component of the BMP-2 cascade, directly governing Osterix expression by maintaining AKT phosphorylation levels; its depletion results in the severe impairment of the mineralization program [48] . Meanwhile, SNHG13 (DANCR) suppresses osteogenic differentiation via a nucleic acid-independent mechanism, directly binding FOXO1 and inducing its ubiquitin-proteasomal degradation [46] .
Although these mechanisms illuminate the multifaceted potential of lncRNAs, the current chain of evidence faces two formidable challenges. First is the issue of model specificity versus universality: the epigenetic mechanism of SNHG3 was primarily established in cardiovascular valve calcification models, while lnc-OAD validation remains confined to in vitro assays. Given the unique mechanical and metabolic signatures of the bone microenvironment, the reproducibility of these mechanisms must be verified in orthotopic animal models of OP or osseous defects. Second, transcript heterogeneity constitutes a cryptic barrier to functional resolution. Distinct splice variants of SNHG2 (GAS5) have been shown to exert diametrically opposed biological effects [46] , implying that traditional whole-gene loss-of-function approaches may obscure the authentic roles of specific transcripts. Future intervention strategies must advance toward precision splicing modulation, utilizing systems like CRISPR-Cas13d to target exon junctions. This will enable the point-to-point regulation of specific transcript isoforms, moving beyond broad-spectrum gene silencing.
Targeted Treatment Strategies for OP
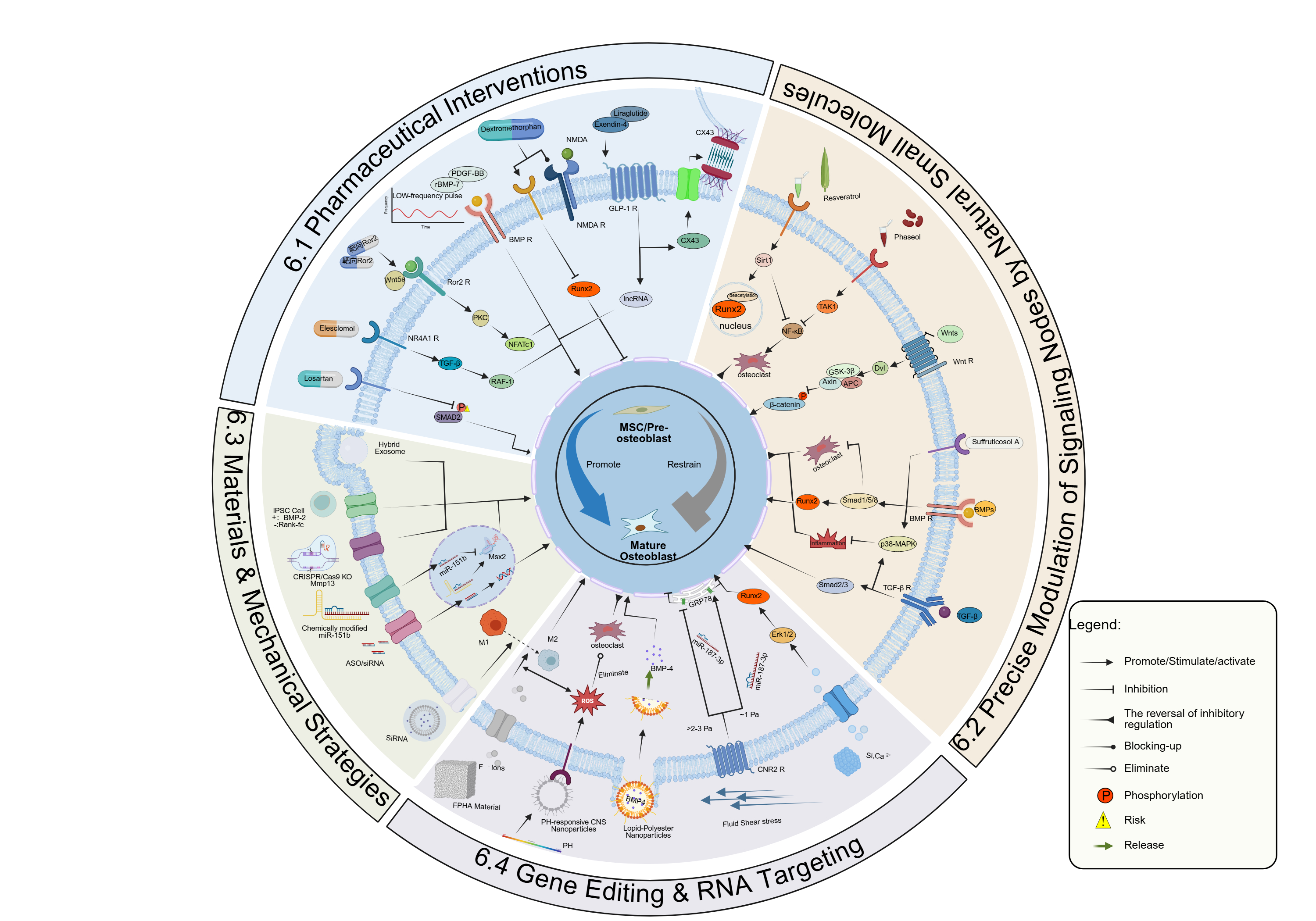
A schematic overview of the targeted intervention landscape—spanning pharmacological agents, natural small molecules, materials/mechanical strategies, and gene/RNA targeting—within the osteogenic regulatory network is presented in Figure 3.
Figure 3. The four-dimensional landscape of precision therapeutic
strategies targeting the osteogenic regulatory network.
This radial diagram synthesizes next-generation intervention paradigms,
categorizing strategies into four precision dimensions that converge to
restore the homeostatic MSC-to-osteoblast differentiation trajectory
(Center). (6.1) Pathology-Dependent Pharmacological Reprogramming (Top
Left): Moving beyond broad-spectrum agents, this quadrant highlights
context-specific modulation. Losartan acts as a non-canonical protector
to rectify hyperactive TGF-β signaling in OI, whereas Elesclomol functions
as an agonist to reactivate dormant pathways via NR4A1. The schematic
also emphasizes "spatiotemporal rhythm," illustrating how low-frequency
pulsatile delivery of BMP-7 (inset graph) prevents receptor
desensitization, contrasting with the risks of continuous dosing.
Notably, the yellow hazard icon warns against collateral damage, such as
Dextromethorphan-induced Runx2 suppression. (6.2) Natural Small Molecule
Node Modulation (Top Right): Phytochemicals serve as precise allosteric
modulators. Phaseol binds the TAK1 hub, redirecting signaling flux from
the inflammatory NF-κB cascade to the osteogenic p38/BMP pathway.
Resveratrol activates the deacetylase SIRT1, which stabilizes Runx2
post-translationally to bypass inflammatory suppression. Suffruticosol A
synergistically reactivates canonical and non-canonical BMP signaling.
(6.3) Biomaterial and Mechanical Engineering (Bottom): Constructing a
biomimetic microenvironment. Strategies include FPHA biomaterials that
drive immunomodulatory M2 macrophage polarization and pH-responsive CNS
nanoparticles that scavenge ROS in the acidic osteoporotic niche. The
mechanotransduction pathway demonstrates the "biphasic window," where
fluid shear stress (~1 Pa) activates the CNR2 receptor to promote
anabolism, whereas excessive force (>2-3 Pa) induces damage.
(6.4) Gene Editing and RNA Targeting (Bottom Left):
Genetic source reprogramming.
This dimension utilizes Hybrid Exosomes to penetrate the mineralized
matrix barrier for CRISPR/Cas9 delivery, enabling "scarless" repair.
Concurrently, ASOs and chemically modified miRNA inhibitors provide
precise post-transcriptional silencing to derepress osteogenic factors
like Msx2.
With the deepening understanding of the multidimensional regulatory networks governing osteogenic differentiation, the treatment of OP should transition from traditional approaches such as hormone replacement or broad-spectrum anti-resorptive therapies toward precise modifications targeting critical signaling nodes, microenvironmental homeostasis, and genetic information.
Pharmaceutical Interventions
Consequently, next-generation pharmacological strategies are moving beyond the binary paradigm of simple pathway activation or blockade. Instead, the focus has shifted toward bidirectional fine-tuning and spatiotemporal optimization tailored to specific pathological states.
Pathology-Dependent Bidirectional Regulation:
The TGF-β signaling pathway exhibits a dichotomous nature, manifesting diametrically opposed roles across different stages of bone pathology. This necessitates a "divide-and-conquer" strategy for precision intervention.
In conditions driven by aberrant signaling and matrix disarray, such as OI, the angiotensin II receptor antagonist Losartan exerts a non-canonical osteoprotective function. By specifically curbing the aberrant phosphorylation of intracellular SMAD2 and ligand secretion, Losartan has been shown to ameliorate cortical bone microarchitecture [49] . Conversely, in scenarios of differentiation arrest characterized by signaling deficiency, the anticancer agent Elesclomol functions as an agonist for the orphan nuclear receptor NR4A1. By reconstructing the "NR4A1-TGF-β-RAF-1" cascade, it functions to reactivate the dormant osteogenic program [50] . However, such interventions must be vigilant against off-target effects. For instance, while the chemotherapeutic Bleomycin suppresses tumors, it paradoxically induces SMAD7 expression, thereby blocking Runx2 activity and potentially contributing to iatrogenic bone loss [28] .
Non-canonical Pathways and Spatiotemporal Kinetics: To circumvent the tumorigenic risks associated with canonical signaling and enhance therapeutic efficiency, the exploitation of non-canonical pathways and the optimization of pharmacokinetics constitute a critical pivot point.
In myeloma-associated bone disease, targeting Ror2-mediated non-canonical Wnt signaling may offer a more favorable safety profile. By activating the Wnt5a/PKC/NFATc1 axis, this approach precisely counteracts the osteogenic inhibition inherent to the tumor microenvironment without perturbing oncogenic networks [12] . Spatiotemporal Rhythm: In combinatorial therapy, the superposition of growth factors is not a matter of simple arithmetic but a rigorous calculation of "spatiotemporal rhythm." The synergistic efficacy of recombinant human BMP-7 and PDGF-BB is strictly contingent upon a low-frequency pulsatile regimen; continuous high-frequency stimulation paradoxically induces receptor desensitization or signal antagonism, leading to ectopic ossification [10] . This discovery profoundly revises conventional drug administration paradigms. Future interventions must incorporate smart bioinspired carriers that mimic physiological pulsatile release rhythms, in order to develop strategies for mitigating off-target effects.
The Opportunities and Complexity of Drug Repurposing: The "drug repurposing" of systemic agents has significantly expanded the landscape of osteogenic intervention, yet it introduces the complexity of cross-system regulation.
Metabolic agents, specifically GLP-1 receptor agonists, have been confirmed to ameliorate the bone microenvironment by enhancing Cx43 gap junctions or mediating lncRNA-driven epigenetic derepression [45,51] . In stark contrast, the neurological agent Dextromethorphan, while blocking NMDA receptors, significantly suppresses Runx2 expression as a form of "collateral damage" [52] . Given the universality of signaling pathways across tissues, existing repurposing strategies urgently require the establishment of robust bone safety assessment frameworks. When utilizing non-orthopedic drugs for chronic conditions, it is imperative to synchronously monitor bone metabolic indices or develop bone-targeted delivery systems to mitigate the risk of off-target skeletal toxicity.
Precision Modulation of Signaling Nodes by Natural Small Molecules
Natural products represent more than mere nutritional supplements; they constitute a vast chemical library for identifying high-selectivity modulators of signaling pathways. Distinct from strategies relying on crude extracts, phytochemical small molecules with defined structures function as allosteric modulators or enzymatic inhibitors to precisely intervene at critical kinase nodes and epigenetic enzymes within the osteogenic network. This inherent "structural diversity" offers unique solutions to circumvent the off-target toxicities often associated with conventional synthetic pharmacotherapies.
Redirection of Kinase Nodes: Targeting the TAK1 Hub As mentioned earlier, TAK1 serves as a pivotal crossroad integrating inflammatory signals with osteogenic cues. Within the chronic inflammatory microenvironment of OP, Phaseol (also known as Phaseollin) has been identified as a natural small molecule capable of specifically binding and modulating TAK1 activity. Rather than simply blockading TAK1, Phaseol suppresses the hyperactivation of the downstream NF-κB cascade, effectively redirecting the signaling flux toward the p38 MAPK and Smad1/5/8 pathways to favor osteogenesis [53] . This strategy of "redirection rather than occlusion" effectively converts inflammatory stress within the microenvironment into an osteogenic driver, providing a paradigm for the precise management of inflammatory bone loss.
Maintenance of Homeostasis via Epigenetic and Post-Translational Modifications: Beyond the direct activation of signaling pathways, natural small molecules enhance osteogenic efficacy by modulating the post-translational modifications (PTMs) of transcription factors. Resveratrol, a specific agonist of Sirtuin 1 (SIRT1), functions through a deacetylation mechanism. Research demonstrates that activated SIRT1 directly deacetylates Runx2, protecting it from ubiquitin-proteasome degradation. This significantly enhances the protein stability and transcriptional activity of Runx2 without necessitating an increase in its mRNA levels [54] . Furthermore, SIRT1 suppresses NF-κB acetylation, thereby alleviating the inhibition of osteogenesis by inflammatory cytokines. This dual mechanism suggests that targeting deacetylases is a potent strategy for reversing the "functional silence" of transcription factors in senile OP.
Synergistic Activation and Translational Perspectives: Addressing the issues of resistance or compensation associated with single-pathway targeting, certain natural molecules exhibit synergistic multi-target effects. Suffruticosol A has been confirmed to synchronously activate both the canonical BMP2/Smad1/5/8 pathway and the non-canonical MAPK cascade, reigniting the differentiation program under stress-induced suppression [55] . However, clinical translation is currently hampered by poor bioavailability and target specificity drift. Future research must transcend simple phenotypic screening; rather, these molecules should be regarded as lead compounds. By integrating Structure-Based Drug Design (SBDD), future efforts should aim to develop next-generation small molecules that combine high bone tissue affinity with precise molecular targeting (Table 1).
Table 1. Representative Natural Small Molecules Targeting Osteogenic Signaling Nodes
|
Compound |
Classification |
Primary Molecular Target |
Mechanism of Action (Signaling Modulation) |
Reference |
|
Phaseol |
Coumestan |
TAK1 (Kinase Hub) |
Allosterically modulates TAK1 to inhibit NF-κB signaling while redirecting flux to p38 MAPK/BMP pathways. |
[53] |
|
Resveratrol |
Polyphenol |
SIRT1 (Deacetylase) |
Activates SIRT1 to deacetylate Runx2, preventing its proteasomal degradation; concurrently inhibits NF-κB acetylation. |
[54] |
|
Suffruticosol A |
Stilbene Oligomer |
BMP / MAPK |
Synergistically activates canonical Smad1/5/8 and non-canonical MAPK cascades to rescue differentiation arrest. |
[55] |
Note: Summary of representative natural small molecules and their molecular mechanisms in driving the osteogenic differentiation program via precise modulation of critical signaling hubs and remodeling of intracellular signaling fluxes.
Materials & Mechanical Strategies
The core philosophy of material and mechanical strategies lies in constructing a biomimetic local microenvironment. By leveraging physicochemical cues as "solid-state signaling sources" that transcend the cell membrane, these approaches effectively circumvent the targeting limitations inherent to systemic drug administration.
Osteoimmunomodulation: Modern biomaterial design has evolved beyond the mere activation of integrin signaling via nanotopography [56] toward the active remodeling of the immune microenvironment—a concept known as osteoimmunomodulation. Inorganic ions have been validated as independent signaling inducers; functionalized scaffolds releasing silicon and calcium ions are capable of promoting osteogenic differentiation via the Erk1/2-Runx2 axis, reducing the reliance on exogenous growth factors [57] . Material-mediated metabolic reprogramming represents a novel therapeutic frontier. Fluorine-doped hydroxyapatite (FPHA), by releasing trace fluoride, switches macrophage metabolism from pro-inflammatory glycolysis to oxidative phosphorylation (OxPhos), thereby fostering an M2-phenotype-dominant, pro-osteogenic microenvironment [58] . Addressing the acidic pathological milieu of OP, pH-responsive cerium nanoparticles (CNS) sense local acidity and generate ROS in situ, achieving selective depletion of mature osteoclasts while preserving osteogenic precursors [59] . Current strategies predominantly focus on singular functions, failing to address the complex pathological cascade of OP. Future intelligent materials should aim to construct "logic gate-like" multi-stage response systems. These would integrate sequential functionalities—"early osteoclast clearance → intermediate immune tolerance → late bone regeneration"—within a single vector.
Biochemical Factors: Kinetics and Intracellular Delivery: The therapeutic efficacy of biochemical factors is not dictated solely by dosage but is strictly governed by their release kinetics. Evidence confirms that mimicking physiological sustained release is critical for initiating anabolism, whereas high-dose burst release is not only ineffective but may precipitate catabolism or systemic adverse events [26] . To resolve this paradox, nanodelivery technology is evolving from simple physical encapsulation to "intracellular programmability." Utilizing a dual mode of "extracellular receptor binding + intracellular degradation," lipid-polyester nanoparticles have shown the potential to achieve efficient intracellular delivery and sustained release of BMP4 [60] . Most controlled-release systems are evaluated under static in vitro conditions, neglecting the dynamic washout effects of body fluid flow. Consequently, translational research must incorporate microfluidic chip technology to simulate the physiological hemodynamic environment, thereby calibrating the in vivo degradation rates and release kinetics of nanocarriers to improve the translational predictive value.
Physical Therapy: The Biphasic Window of Mechanotransduction: The essence of physical therapy lies in the precision control of mechanical stimulus intensity. Fluid shear stress regulates osteogenic metabolism through a strict "biphasic window": it promotes anabolism only within a specific intensity range (~1 Pa), whereas exceeding this threshold (>2-3 Pa) converts the stimulus into a damage signal [61] . This mechano-chemical transduction is highly dependent on intracellular precision sensors, including the ER chaperone GRP78 and the miR-187-3p/CNR2 axis [44,62] . These sensors function as "molecular switches," influencing whether physical signals activate osteogenic pathways or induce apoptosis. Existing rehabilitation strategies often employ "one-size-fits-all" mechanical parameters, ignoring the critical fact that the mechanosensitivity of cells in osteoporotic patients is likely altered. Future research should prioritize the development of companion diagnostic tools based on mechanosensitive markers. These would serve as the basis for customizing implant design parameters and postoperative rehabilitation protocols tailored to the patient's specific mechanobiological profile.
Gene Editing & RNA Targeting
Distinct from pharmacological and material interventions that operate downstream at the protein or cellular level, gene editing and RNA technologies aim to execute "precise reprogramming" directly at the genetic source, offering promising strategies for treating hereditary bone diseases and managing refractory OP.
At the post-transcriptional modification level, RNA-targeting strategies are considered to possess considerable potential for high specificity. Research on OI indicates that employing antisense oligonucleotides (ASOs) or siRNA to achieve "allele-specific silencing" can potentially correct the haploinsufficiency [63,64] . This precise silencing mechanism extends to the non-coding RNA regulatory network; for instance, chemically modified miR-151b inhibitors specifically relieve the repression of Msx2, thereby effectively restoring the transcriptional activity of osteogenic genes [43] . Concurrently, genome editing technology is undergoing a paradigm shift from "gene disruption" to "precision repair." While CRISPR/Cas9-mediated knockout of Mmp13 has proven effective in delaying osteoarthritis progression [65] , the field is accelerating toward Base Editing and Prime Editing. Theoretically, these novel technologies may mitigate the safety concerns of genomic instability associated with traditional editing by bypassing the reliance on DNA double-strand breaks (DSBs), thus enabling the "scarless repair" of point mutations [63] . Furthermore, "ex vivo gene enhancement" combined with iPSCs represents an alternative translational pathway. By re-infusing cells genetically modified to overexpress BMP-2 or knock down RANK-Fc, this approach achieves a synergistic potentiation of "cellular regeneration" and "genetic regulation" [66,67] .
Despite clear therapeutic targets, the clinical translation of nucleic acid drugs has long been impeded by the instability of naked moieties and biological membrane barriers. Current delivery strategies are progressively pivoting away from viral vectors, which carry immunogenic risks, toward non-viral vectors dominated by Lipid Nanoparticles (LNPs) and functionalized biomaterials. These alternatives leverage superior biocompatibility and modification flexibility to shield the nucleic acid payload [68] . Addressing the unique barrier presented by the dense extracellular matrix (ECM) of bone tissue, engineered "hybrid exosomes" have presented a promising approach for delivering CRISPR systems via membrane fusion, facilitating the penetration of macromolecular drugs into the bone/cartilage matrix [65] . At the cellular targeting level, nanocarrier-mediated siRNA delivery has achieved specific modulation of macrophages within the osteoimmune microenvironment, opening novel avenues for ameliorating the osteogenic niche [69] .
Currently, while gene therapy exhibits vast prospects, the critical leap from "bench to bedside" hinges on the establishment of rigorous evaluation and quality control frameworks. Confronting potential off-target effects and long-term safety concerns, future translational research must not be satisfied solely with numerical improvements in editing efficiency. Instead, it is imperative to construct functional evaluation systems based on gold-standard markers such as Runx2 and COL1A1 [70] . This must be coupled with Whole Genome Sequencing (WGS) to comprehensively screen for off-target risks, facilitating the translation of precise modulation at the molecular level translates into functional benefits in histological bone regeneration, rather than merely transient perturbations in signaling pathways.
Future Perspectives
The therapeutic paradigm for OP is undergoing a fundamental shift from a singular reliance on anti-resorptive strategies toward "precision homeostatic remodeling" grounded in systems biology. This evolution is predicated on a profound comprehension of metabolic regulation mechanisms: key signaling pathways, such as Wnt and BMP, exhibit high context-dependency, with their biological effects rigorously contingent upon the specific microenvironment. This context-sensitivity elucidates a primary failure mode of traditional single-target therapies under pathological conditions. Central to systemic bone loss is the dysregulated competition between the Runx2-driven osteogenic and PPARγ-driven adipogenic lineages. This imbalance is orchestrated by multidimensional regulation, notably progenitor pool exhaustion precipitated by Wnt10b attenuation and epigenetic silencing mediated by the NLK-SETDB1 complex. Concurrently, non-coding RNAs modulate differentiation thresholds predominantly via a logic of "disinhibition," functioning by dismantling endogenous checkpoints such as SOCS3.
Corresponding translational trajectories have crystallized into three distinct avenues. First, the design of "functionally decoupled" pharmacotherapies aims to precisely delineate and separate the osteo-anabolic benefits of signaling pathways from their deleterious off-target effects. Second, the development of smart, responsive biomaterials seeks to engineer chronologically orchestrated therapies that achieve "initial osteoclast inhibition followed by osteogenic induction." Third, the advancement of "scarless" gene editing—integrating Base Editing and Prime Editing with exosome-based delivery—offers a strategy to correct genetic defects while precluding the generation of hazardous DNA DSBs.
Nevertheless, the "bench-to-bedside" translation currently confronts three critical bottlenecks: the paucity of efficient delivery vectors capable of penetrating the dense mineralized matrix barrier of bone tissue; the lacuna in long-term safety evaluation frameworks, particularly regarding the surveillance of osteogenic hyper-activation and off-target editing risks; and the limitations of extant disease models, which necessitate the adoption of humanized organoids and large animal models to validate clinical predictive efficacy. Future breakthroughs will likely rely on overcoming a rigorous and fortified bridge spanning the chasm between these frontier mechanistic insights and the pragmatic challenges of clinical translation.
Abbreviations
adipose-derived stem cells: ASCs; alkaline phosphatase: ALP; bone mineral density: BMD; Bone Morphogenetic Protein: BMP; bone marrow stromal cells: BMSCs; Cyclophilin A: CypA; competing endogenous RNA: ceRNA; dextromethorphan: DXM; Frizzled: FZD; fluid shear stress: FFSS; growth response protein 1: EGR1; glucagon-like peptide-1 receptor: GLP-1R; Long non-coding RNAs: lncRNAs; lipopolysaccharide: LPS; lipid nanoparticle: LNP; mesenchymal stem cells: MSCs; microRNAs: miRNAs; mature osteoclasts: mOCs; Notch intracellular domain: NICD; Osteoporosis: OP; osteoporotic adipose-derived stem cells: OP-ASCs; osteopontin: OPN; Oncostatin M receptor: OSMR; osteogenesis imperfecta: OIM; osteocalcin: OCN; ovariectomized: OVX; protein kinase C: PKC; Phaseolamin: PHA; precursor osteoclasts: pOCs; recombinant human BMP7: rhBMP7; reactive oxygen species: ROS; SET domain bifurcation histone lysine methyltransferase 1: SETDB1; soy peptides: SOP; trabecular number: Tb.N; T-cell factor: TCF; type I collagen alpha 1: COL1A1; Transforming Growth Factor-beta: TGF-β; traumatic brain injury: TBI; umbilical cord mesenchymal stem cells: UCMSCs; World Health Organization: WHO; 3' untranslated regions: 3'UTRs.
Declarations
Author contributions
Chuntao Liang: Writing–review & editing, Writing–original draft. Dongcheng Peng: Writing–review & editing, Validation. Hongkai Wang: Writing–review & editing, Conceptualization, Supervision. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Funding information
Not applicable.
Ethics approval and consent to participate
Not applicable.
Competing Interests
The authors declare that they have no existing or potential commercial or financial relationships that could create a conflict of interest at the time of conducting this study.
Data Availability
Not applicable.
References
[6].Włodarski KH. Properties and origin of osteoblasts. (1990). Clinical Orthopaedics and Related Research. (252):276–93.
[64]Morello R. Osteogenesis imperfecta and therapeutics. (2018). Matrix Biology. 71–72:294–312.
[69].Song S, Xia H, Guo M, Wang S, Zhang S, Ma P, et al. Role of macrophage in nanomedicine-based disease treatment. (2021). Drug Delivery.
 Figures
Figures References
References Peer
Peer Information
Information
Figure 1. The multidimensional signaling topology and hierarchical
integration of the osteoblast differentiation network.
This schematic illustrates the systems-level logic governing osteogenic
fate, highlighting the transition from extracellular signal integration to
nuclear transcriptional reprogramming and epigenetic locking. (A) Membrane
Reception and Context-Dependent Crosstalk: The network initiates with the
spatiotemporal integration of core signals. Canonical Wnt signaling (left)
is triggered by ligands such as Wnt10b and Wnt3a, which stabilize β-catenin
to drive progenitor maintenance. In contrast, non-canonical Wnt5a exhibits
profound "context-dependency": it engages the Ror2/FZD receptor complex to
activate the osteogenic PKC and CaMKII axes, while acting via a paracrine
mechanism on osteoclast precursors (top right) to upregulate RANK and
amplify resorption. Meanwhile, the BMP and TGF-β pathways (in the middle)
not only activate their respective pathways but also converge at the MAPK
effector factor, among which the BMP2-activated pathway is strictly
regulated by auxiliary sensors (such as GPRC5C), which is epigenetically
derepressed upon the removal of EZH2-mediated H3K27me3 marks. (B)
Cytoplasmic Convergence Hubs: Distinct signals converge on TAK1, a pivotal
kinase nexus that integrates inputs from Wnt (via CaMKII), BMP/TGF-β, and
inflammatory cytokines (TNF-α/IL-1) to prioritize cell survival and
differentiation. Signal flux is further fine-tuned by intracellular
feedback loops: EGR1 acts as a "cytosolic adapter" bridging ERK to Smad
activation (positive feedback), whereas OSMR functions as a metabolic
brake by suppressing ERK phosphorylation and blocking autophagic flux
(indicated by LC3II reduction and P62 accumulation). (C) The Runx2-PPARγ
Equilibrium and Epigenetic Blockade: In the nucleus, Runx2 serves as the
master transcriptional hub, synergized by co-factors and self-reinforced
by repressing the antagonist CHRDL1. The ultimate lineage commitment
hinges on the reciprocal equilibrium between Runx2 and the adipogenic
regulator PPARγ. The diagram highlights a critical pathological
mechanism: the collapse of this balance is prevented by a Wnt5a-induced
epigenetic blockade, where the NLK-CHD7-SETDB1 complex catalyzes H3K9
methylation at the PPARγ promoter, thereby silencing adipogenesis to
consolidate the osteogenic program.
Figure 2. The post-transcriptional logic and epigenetic
architecture of non-coding RNA-mediated osteogenic fate determination.
This schematic delineates the dichotomous regulatory landscape governed by
non-coding RNAs, contrasting the logic of "disinhibition" (Left, Blue
panel) against "suppression" (Right, Orange panel) in the control of
osteoblast differentiation. (A) The Osteo-Anabolic Network: Disinhibition
and Epigenetic Derepression. The left panel illustrates how osteogenic
signals are safeguarded through the neutralization of endogenous
inhibitors. Signaling Disinhibition: MicroRNAs act as checkpoints to
release signaling blockades. miR-218-5p targets the inhibitor SOCS3,
thereby unleashing Smad1 activity; miR-92a-3p, triggered by TBI stress,
suppresses IBSP to reactivate the PI3K/AKT axis; and mechanical stretch
downregulates miR-187-3p, relieving the repression on the CNR2 receptor.
ceRNA & Epigenetic Scaffolds: Long non-coding RNAs (lncRNAs) function
through different structural mechanisms. In the cytoplasm, SNHG14 and
LINC00968 act as competitive endogenous RNAs to protect the transcripts
of AKT2 and Runx2 by inhibiting miR-2861 and adsorbing miR-3658,
respectively. In the nucleus, SNHG3 acts as an epigenetic decoy, anchoring
EZH2 (of the PRC2 complex) to prevent H3K27me3 deposition at the BMP2
promoter, thus maintaining transcriptional accessibility. (B) The
Osteo-Suppressive Network: Feedback Paradox and Degradation. The right
panel depicts mechanisms leading to differentiation arrest. The ceRNA
Paradox: Highlighting functional divergence, SNHG1 adsorbs miRNAs such as
miR-181c-5p and miR-101, preventing the inhibition of Wnt pathway
antagonists DKK1 and SFRP1 (thereby stabilizing them), and ultimately
leading to the blocking of the classical Wnt signaling pathway. Targeted
Degradation: SNHG13 operates via a nucleic-acid-independent mechanism,
directly binding FOXO1 to induce its ubiquitin-proteasomal degradation.
Concurrently, miR-151b clamps the 3'UTR of Msx2, severing the downstream
transcriptional cascade essential for mineralization.
Figure 3. The four-dimensional landscape of precision therapeutic
strategies targeting the osteogenic regulatory network.
This radial diagram synthesizes next-generation intervention paradigms,
categorizing strategies into four precision dimensions that converge to
restore the homeostatic MSC-to-osteoblast differentiation trajectory
(Center). (6.1) Pathology-Dependent Pharmacological Reprogramming (Top
Left): Moving beyond broad-spectrum agents, this quadrant highlights
context-specific modulation. Losartan acts as a non-canonical protector
to rectify hyperactive TGF-β signaling in OI, whereas Elesclomol functions
as an agonist to reactivate dormant pathways via NR4A1. The schematic
also emphasizes "spatiotemporal rhythm," illustrating how low-frequency
pulsatile delivery of BMP-7 (inset graph) prevents receptor
desensitization, contrasting with the risks of continuous dosing.
Notably, the yellow hazard icon warns against collateral damage, such as
Dextromethorphan-induced Runx2 suppression. (6.2) Natural Small Molecule
Node Modulation (Top Right): Phytochemicals serve as precise allosteric
modulators. Phaseol binds the TAK1 hub, redirecting signaling flux from
the inflammatory NF-κB cascade to the osteogenic p38/BMP pathway.
Resveratrol activates the deacetylase SIRT1, which stabilizes Runx2
post-translationally to bypass inflammatory suppression. Suffruticosol A
synergistically reactivates canonical and non-canonical BMP signaling.
(6.3) Biomaterial and Mechanical Engineering (Bottom): Constructing a
biomimetic microenvironment. Strategies include FPHA biomaterials that
drive immunomodulatory M2 macrophage polarization and pH-responsive CNS
nanoparticles that scavenge ROS in the acidic osteoporotic niche. The
mechanotransduction pathway demonstrates the "biphasic window," where
fluid shear stress (~1 Pa) activates the CNR2 receptor to promote
anabolism, whereas excessive force (>2-3 Pa) induces damage.
(6.4) Gene Editing and RNA Targeting (Bottom Left):
Genetic source reprogramming.
This dimension utilizes Hybrid Exosomes to penetrate the mineralized
matrix barrier for CRISPR/Cas9 delivery, enabling "scarless" repair.
Concurrently, ASOs and chemically modified miRNA inhibitors provide
precise post-transcriptional silencing to derepress osteogenic factors
like Msx2.
[6].Włodarski KH. Properties and origin of osteoblasts. (1990). Clinical Orthopaedics and Related Research. (252):276–93.
[64]Morello R. Osteogenesis imperfecta and therapeutics. (2018). Matrix Biology. 71–72:294–312.
[69].Song S, Xia H, Guo M, Wang S, Zhang S, Ma P, et al. Role of macrophage in nanomedicine-based disease treatment. (2021). Drug Delivery.
Peer-review Terminology
Identity transparency: Single anonymized
Reviewer interacts with: Editor
Details
This is an open access article under the terms of the Creative Commons Attribution License(http://creativecommons.org/licenses/by/4.0/), which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Publication History
Received 2025-12-01
Accepted 2026-01-29
Published 2026-03-09