




Abstract

Objective:Two patients from the same family with glucose transporter type 1 deficiency syndrome (GLUT1-DS) were reported, and they exhibited markedly different clinical symptoms.
Methods:We described two patients with GLUT1 deficiency syndrome (GLUT1-DS) and compared their clinical presentations.
Results:Two patients from the same family have the same genetic mutation but exhibit vastly different clinical presentations.
Conclusion: GLUT1-DS presents complex and varied clinical manifestations, complicating diagnosis and requiring multiple diagnostic methods. Treatment options are limited, with the ketogenic diet as a potential therapeutic approach.
Keywords:GLUT1DS, seizures, ketogenic diet, prognosis, SLC2A1
Introduction
Glucose Transporter Type 1 Deficiency Syndrome (GLUT1-DS) is a rare genetic disorder, first reported in 1991[1] . This condition is predominantly caused by pathogenic variants in the SLC2A1 gene that encodes glucose transporter type 1 (GLUT1). This genetic alteration impair glucose transport across the blood-brain barrier, resulting in neurological manifestations associated with brain energy deficit. Typical clinical manifestations of GLUT1-DS include delayed motor and cognitive development, infantile seizures, deceleration of head growth (often resulting in acquired microcephaly), and movement disorders (accompanied by ataxia, muscle tone abnormalities, and spasms)[2] . About 90% of GLUT1-DS cases are caused by de novo mutations, and this disease is rarely inherited in an autosomal dominant or recessive manner. Therefore, most patients have no family history of the disease. Heterozygous parents may present with a benign phenotype or be entirely asymptomatic[3] .
In GLUT1-DS patients, glucose and lactate levels in the cerebrospinal fluid are lower than normal. Therefore, in addition to confirming the presence of mutations through genetic testing, the cerebrospinal fluid/serum glucose ratio is also an important clinical diagnostic indicator for GLUT1-DS (normal GLUT1-DS <0.6, classic presentation <0.35)[4] . The ketogenic diet can provide alternative fuel for the brain. Early use of the ketogenic diet in treatment can improve or even reverse symptoms[5] .
In this study, we report two specific cases where a father and son, sharing the same mutation site, exhibit different manifestations.
Presentation of Case
Case
A 2-year-and-9-month-old boy, with unspecified Apgar scores, born from a first pregnancy and delivery, delivered at full term weighing 3250 grams, had no history of asphyxia, hypoxia, or resuscitation at birth. At four months of age, the child began experiencing seizures manifested as a fixed gaze with a "blank stare" lasting several seconds, or head turning from side to side accompanied by synchronous eye movements. These episodes persisted for 1-2 minutes and occurred 2-3 times every two months. At 5 months of age, the child exhibits isolated episodes of raising both hands or twitching of both lower limbs, with each episode lasting about 1-2 seconds and resolving on its own. The frequency of these episodes is 3-5 times per day, occurring every few days, with the longest period without such episodes being 2 weeks. At 9 months of age, the episodes become frequent, occurring up to a dozen times a day, with a frequency of every 3-5 days. At 11 months of age, dynamic electroencephalography (EEG) indicated widespread spike-and-slow-wave activity, slow waves, and slow waves mixed with spikes during sleep. Myoclonic seizures were observed, leading to a diagnosis of epilepsy. After 1 week of oral sodium valproate treatment, seizures recurred. At 1 year and 7 months old, following a pulmonary infection, the child began to exhibit head turning to the left and right, with both eyes following the head movements, lasting approximately 1-5 minutes. This occurred several times over a 2-week period, with a normal routine EEG. Continue treatment with sodium valproate. The patient's family sought further treatment, and the child was re-examined at our hospital at 1 year and 11 months of age. Completion of the GESELL developmental scale indicated mild developmental delay. Fasting blood glucose was normal, and abdominal and urogenital ultrasound showed no significant abnormalities. MRI of the head with contrast revealed bilateral choroid plexus cysts, while the rest of the brain MRI scan, both plain and with contrast, showed no obvious abnormalities. At 2 years and 6 months, a lumbar puncture in the child revealed a low cerebrospinal fluid to serum glucose ratio (1.45/5.05=0.287, <0.35). It is believed that the child's seizures and developmental delay may be related to GLUT-1 deficiency syndrome (GLUT-1DS). It is recommended to complete a lumbar puncture and genetic testing. The patient underwent comprehensive whole-exome genetic testing, which ultimately confirmed GLUT-1 DS caused by the pathogenic mutation in the SLC2A1 gene: p.V69Lfs*21(Figure 1 ).
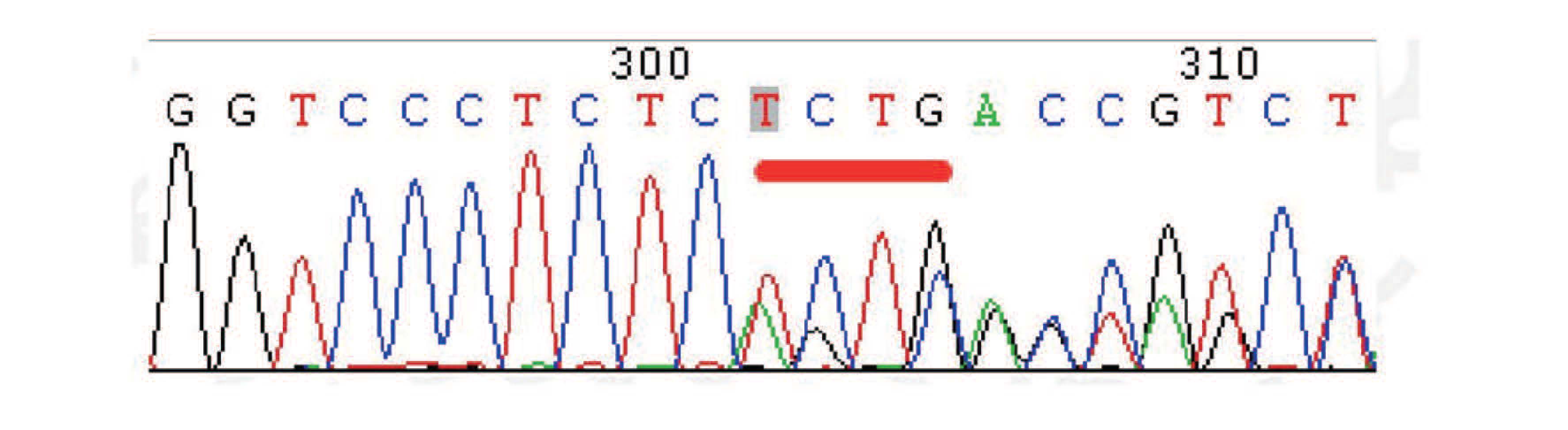
Figure 1. The DNA sequencing results of the patient. The results suggested a heterozygous point mutation (c.940G>A) in exon 7 of the SLC2A1 gene.
Over the past 2 years, the patient has experienced motor and language developmental delays, slightly increased muscle tone in the lower limbs, and multiple episodes of limb jerks or absence seizures. The patient was started on a ketogenic diet at the age of 2 years and 6 months and has been seizure-free since then, with a normal routine EEG on review.
The patient's father, aged 31, had cognitive impairment and delayed reactions since childhood, microcephaly, increased muscle tone, weakness in limbs during hunger, and exhibited a peculiar gait (crossed gait) during prolonged walking. Following the diagnosis of GLUT1-DS in the patient, the father underwent a comprehensive examination. The patient's father was mildly cognitively impaired (MMSE: 26 points, Moca: 20; short-term memory breadth: 8/4, 31 years old with a junior high school education). The 24-hour video EEG showed 3-7 c/s slow wave activity, with no significant changes in the EEG after fasting or eating. At the same time, we performed a lumbar puncture on the patient's father, and the results showed that the cerebrospinal fluid glucose/serum glucose ratio was low (0.38)( Table 1). The genetic testing of the patient's father revealed a mutation in the SLC2A1 gene (p. V69Lfs*21). No obvious abnormalities were found on the head MRI. Other relatives of the patient did not exhibit the mentioned symptoms and did not have the SLC2A1 gene mutation.
Table 1. Cerebrospinal fluid (CSF) examination in both patients.
|
Parameter |
Reference Range |
Changes Typical in GLUT1-DS |
Patient |
Patient’s Father |
|
CSF glucose |
2.5-4.5 mmol/L |
<2.2 mg/dL |
1.45 |
1.85 |
|
Serum lactates |
0.5-2.22mmol/L |
Normal or less than 0.5 |
1.73 |
No Data |
|
CSF/serum glucose ratio |
0.55-0.75 |
0.19-0.59 |
0.287 |
0.378 |
Treatment
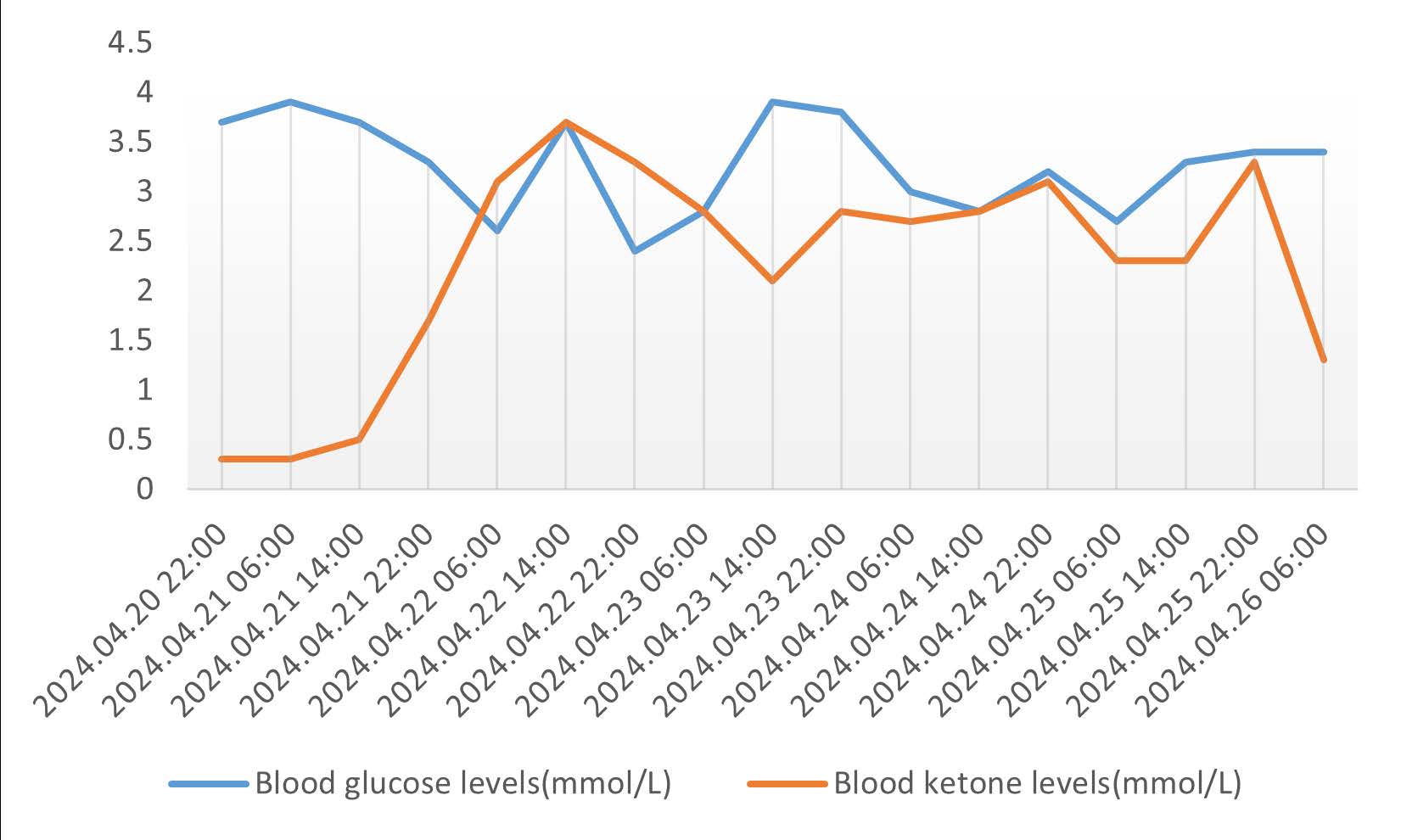
After being diagnosed with GLUT1-DS, the patient started a ketogenic diet with a ratio of 1.5:1 of fat to non-fat components. Sodium valproate was also used concurrently to control epilepsy. Patient's general condition and symptoms vary with fluctuations in serum ketone body levels during the course of treatment. Changes in serum ketone body levels in morning fasting, 2h after lunch and 2h after dinner and changes in blood glucose are shown in Figure 2 .
Figure 2. Changes in the patient's blood glucose and blood ketone levels.
Patients’ Family History
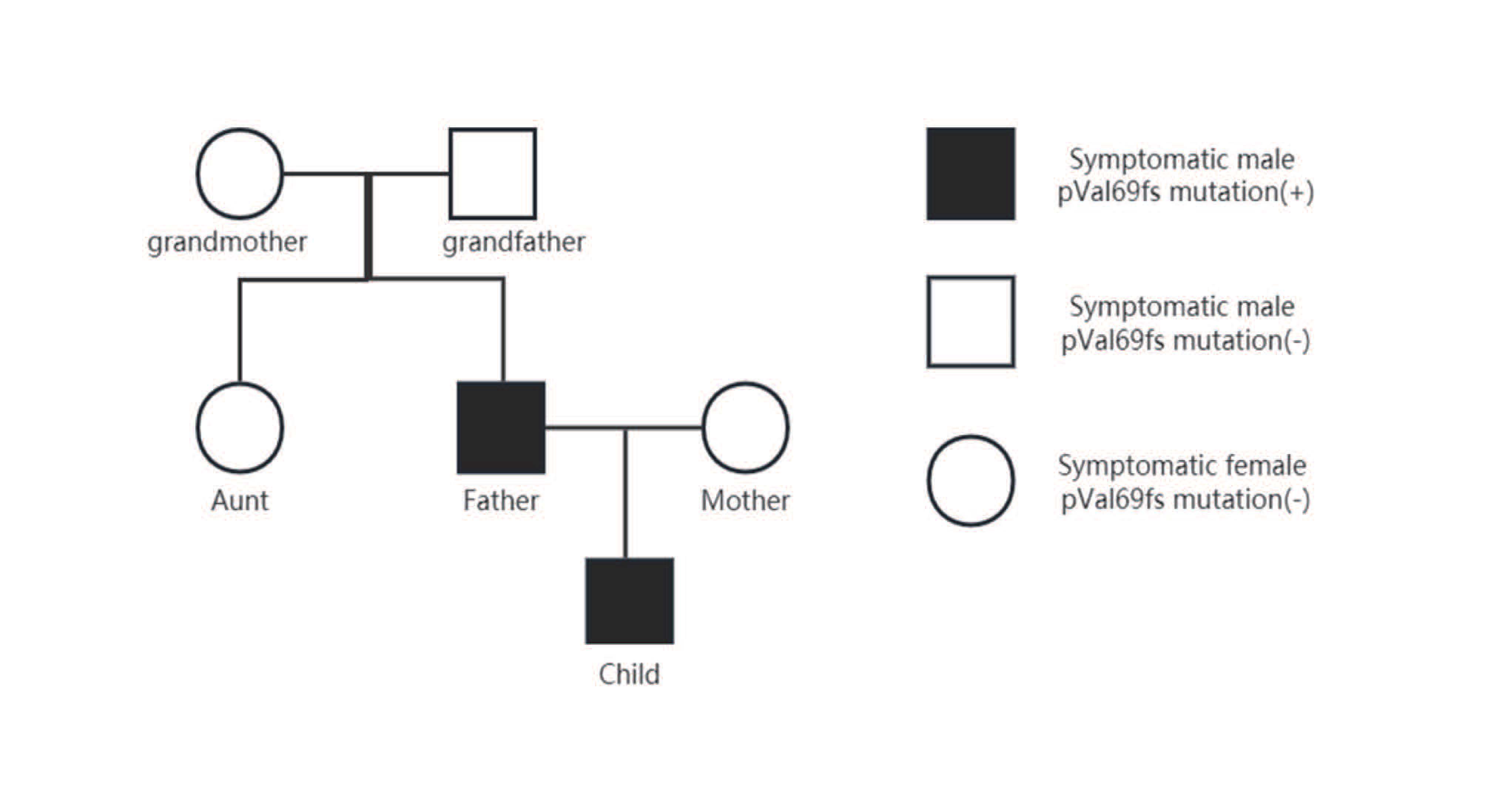
The patient's mother, grandfather, grandmother and aunt also underwent genetic testing for GLUT1-DS. The patient's mother was suspected of having seizures when she was 4 or 5 years old (specific form of seizure not known), and genetic testing was negative, as were results from other family members (Figure 3). To prove that the patient's pathogenic SLC2A1 gene came from their father, and that the father's pathogenic gene originated from a genetic mutation.
Figure 3. Symptoms and the presence of genetic mutation in the family
Discussion
Typical clinical manifestations of GLUT1-DS include seizures, neurodevelopmental delay, dysarthria, acquired microcephaly, and complex movement disorders[6] . Atypical clinical presentations mainly include confusion, drowsiness, sleep disorders, and headaches[3] . Due to its diverse clinical symptoms, diagnosing this condition can be challenging for healthcare professionals. Most patients exhibit clinical symptoms within the first year of life because the brain's glucose metabolism rate is lower during infancy, increases linearly after birth, peaks around age 3, remains elevated for the first decade of life, gradually declines during the second decade after birth, and reaches adult-like glucose utilization rates in early adulthood[7] .
The type of SLC2A1 gene mutation exhibits a gradient correlation with disease severity: Large deletions often lead to severe clinical manifestations, while missense or splice-site mutations typically cause milder symptoms. Nonsense and frameshift mutations generally present with moderate phenotypes. Notably, significant phenotypic variability has been observed among individuals carrying identical pathogenic mutations. For instance, in this cohort, a child demonstrated typical cerebral glucose metabolism disorders, whereas the father carrying the same mutation showed no significant clinical symptoms. Previous studies suggest that differences in mutation proportions caused by somatic mosaicism may be a crucial factor in phenotypic heterogeneity[8] . Additionally, modifier genes regulating glucose transport might participate in phenotype modulation through compensatory mechanisms[9] . The timing of pathogenic mutation occurrence (e.g., mutations arising during later stages of nervous system development) and age-related factors may further contribute to the diversity of clinical manifestations[10] .
Seizures in childhood are a typical feature of GLUT1-DS, but not all patients have seizures, some studies indicate that approximately 10% of patients with GLUT1-DS do not have seizures[11,12] . In this case, the child's father was diagnosed with GLUT1 deficiency syndrome (GLUT1-DS) but does not experience seizures.
Lumbar puncture is an important step in diagnosing GLUT1-DS. Normal blood glucose levels coupled with reduced cerebrospinal fluid (CSF) glucose levels are metabolic hallmarks of GLUT1-DS. Current research indicates that GLUT1-DS patients exhibit CSF glucose levels ranging from 0.9 to 2.9 mmol/L (16.2–52.0 mg/dL), all below 3.3 mmol/L (60 mg/dL), with a CSF glucose-to-blood glucose ratio of 0.19–0.59[13] . Notably, the majority of patients demonstrate CSF glucose levels below 2.2 mmol/L (40 mg/dL) and a CSF glucose-to-blood glucose ratio <0.4. Importantly, the absence of pathogenic variants in SLC2A1 does not exclude a diagnosis of GLUT1-DS[14] . Therefore, despite the invasive nature of lumbar puncture, it is recommended to perform this procedure in all suspected GLUT1-DS cases prior to genetic evaluation.
WES is now the gold standard for diagnosing GLUT1-DS. In pathogenic variants of SLC2A1, approximately 300 nonsense, frameshift, and splice site mutations result in a 50% loss of GLUT1 function, determining the classic phenotype. Conversely, missense mutations that retain 50%-70% of GLUT1 function often lead to mild to moderate phenotypes[15,16] . Of course, comprehensive gathering of family history is also crucial. The presence of related symptoms in the patient's relatives may indicate inheritance of the pathogenic gene from family members, necessitating additional pedigree verification[17] . Moreover, making the correct diagnosis for the patient will also guide doctors in correctly diagnosing the parents of patients who may have been misdiagnosed or undiagnosed. For example, the patient's father in this case. However, not all GLUT1-DS patients have SLC2A1 gene mutations[3] . Some of the more common types of mutations present are Asn34, Gly91, Ser113, Arg126, Arg153, Arg264, Thr295, Arg333, Arg93, Arg212, Gly130, Ala155 and Arg330[16] . In this paper, we report that the patient's mutation site (Val69) was not included in the gnomAD database (East Asia).
During the diagnostic process, the patient undergoes an electroencephalogram (EEG), and in some patients the EEG results may show localised slow-wave activity, high-voltage spikes, or an abnormal spike-wave pattern of discharges[18] . For example, the EEG of the patient's father showed widespread slow wave activity, whereas the patient's EEG during sleep indicated widespread spike-slow waves, slow waves, and slow wave complexes interspersed with spikes. Moreover, an important feature of EEG examinations in GLUT1-DS patients is the normalisation of brain bioelectrical activity as a result of eating[19] . This feature is very helpful for doctors to diagnose the disease.
Currently, in patients with Glut1-DS, the ketogenic diet should be started as early as possible to provide ketone bodies as an alternative energy source for brain cells[4] . There is study reporting that ketogenic diet effectively controls seizures, slows down the progression of movement disorders, and improves cognitive outcomes in children with GLUT1-DS[20] . The ketogenic diet shows clinical effects within 1 week to 6 months, with good tolerability. Discontinuation is rare and linked to metabolic acidosis or GI issues. Early use may boost neurorecovery, especially cognition/language. Response varies: symptom persistence/relapse occurs in some, with efficacy tied to individual metabolism and disease subtypes[21] .
In this case, the patient was placed on a classic ketogenic diet with a fat-to-protein-and-carbohydrate ratio of 4:1. Prior to initiating the diet, a 24-hour fasting induction period was implemented, during which carbohydrate-free fluids were provided while blood glucose and ketone levels were monitored. The ratio was subsequently adjusted based on individual tolerance (range: 2:1 to 4:1). Following the ketogenic diet intervention, the patient experienced cessation of epileptic seizures and demonstrated improvements in both cognitive and motor impairments. The heterogeneity and small scale of our study samples hindered understanding of the relationship between cognitive and psychomotor improvements, seizure status, and dietary types. But long-term use of the ketogenic diet can lead to significant bone loss, cardiovascular atherosclerosis, and even coma[22] . However, compared to the therapeutic effects of the ketogenic diet, its side effects are relatively minor. Therefore, for patients already diagnosed with GLUT1-DS, we recommend early adoption of the ketogenic diet.
Conclusions
When diagnosing refractory epilepsy in children, consideration should be given to GLUT1-DS, especially in cases of epilepsy accompanied by developmental delay and movement disorders. In this study, despite sharing the same genetic mutation, the father and son exhibited different symptoms. Cerebrospinal fluid analysis and genetic testing are crucial for diagnosing GLUT1-DS. Early implementation of the ketogenic diet is crucial for effectively managing the condition in diagnosed patients.[1]
Acknowledgements
Not applicable.
Author contributions
Jiajia Jiao drafted the manuscript and performed EEG recording and analysis; Chengjuan Xie reviewed neurophysiological data and revised the manuscript.
Ethics approval and consent to participate
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines
Funding information
This research was supported by the Open Project of State Key Laboratory of Cognitive Intelligence of China (COGOS-2022003), Natural Science Research Project of Universities in Anhui Province (2022AH051157), Research Fund Project of Anhui Medical University (2022xkj147).
Competing Interests
The authors declare that they have no existing or potential commercial or financial relationships that could create a conflict of interest at the time of conducting this study.
Data Availability
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
References
[1] De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 1991; 325: 703-709. https://doi.org/10.1056/nejm199109053251006.
[2] Anurat K, Khongkhatithum C, Tim-Aroon T, Limwongse C, Thampratankul L. Sleep Disorder: An Overlooked Manifestation of Glucose Transporter Type-1 Deficiency Syndrome. Neuropediatrics 2022; 53: 129-132. https://doi.org/10.1055/s-0041-1736179.
[3] Pawlik W, Okulewicz P, Pawlik J, Krzywińska-Zdeb E. Diagnostic and Clinical Manifestation Differences of Glucose Transporter Type 1 Deficiency Syndrome in a Family with SLC2A1 Gene Mutation. Int J Environ Res Public Health 2022; 19. https://doi.org/10.3390/ijerph19063279.
[4] Klepper J, Akman C, Armeno M, Auvin S, Cervenka M, Cross HJ et al. Glut1 Deficiency Syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open 2020; 5: 354-365. https://doi.org/10.1002/epi4.12414.
[5] Koch H, Weber YG. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav 2019; 91: 90-93. https://doi.org/10.1016/j.yebeh.2018.06.010.
[6] Wang D, Pascual JM, De Vivo D. Glucose Transporter Type 1 Deficiency Syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH et al (eds). GeneReviews(®). University of Washington, Seattle Copyright © 1993-2024, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.: Seattle (WA), 1993
[7] Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep 2013; 13: 342. https://doi.org/10.1007/s11910-013-0342-7.
[8] Takahashi S, Matsufuji M, Yonee C, Tsuru H, Sano N, Oguni H. Somatic mosaicism for a SLC2A1 mutation: implications for genetic counseling for GLUT1 deficiency syndrome. Clin Genet 2017; 91: 932-933. https://doi.org/10.1111/cge.12902.
[9] Sapuppo A, Pavone P, Praticò AD, Ruggieri M, Bertino G, Fiumara A. Genotype-phenotype variable correlation in Wilson disease: clinical history of two sisters with the similar genotype. BMC Med Genet 2020; 21: 128. https://doi.org/10.1186/s12881-020-01062-6.
[10] Jiang Q, Xu H, Yan J, Xu Q, Zheng Y, Li C et al. Sex-specific metabolic alterations in the type 1 diabetic brain of mice revealed by an integrated method of metabolomics and mixed-model. Comput Struct Biotechnol J 2020; 18: 2063-2074. https://doi.org/10.1016/j.csbj.2020.07.019.
[11] Wang D, Pascual JM, DC. DV. Glucose Transporter Type 1 Deficiency Syndrome. . Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A editors Seattle, WA: University of Washington, 1993–2019; 2002 Jul 30 [updated 2018 Mar 1] 2018;
[12] Mullen SA, Suls A, De Jonghe P, Berkovic SF, Scheffer IE. Absence epilepsies with widely variable onset are a key feature of familial GLUT1 deficiency. Neurology 2010; 75: 432-440. https://doi.org/10.1212/WNL.0b013e3181eb58b4.
[13] Pong AW, Geary BR, Engelstad KM, Natarajan A, Yang H, De Vivo DC. Glucose transporter type I deficiency syndrome: epilepsy phenotypes and outcomes. Epilepsia 2012; 53: 1503-1510. https://doi.org/10.1111/j.1528-1167.2012.03592.x.
[14] Yang H, Wang D, Engelstad K, Bagay L, Wei Y, Rotstein M et al. Glut1 deficiency syndrome and erythrocyte glucose uptake assay. Ann Neurol 2011; 70: 996-1005. https://doi.org/10.1002/ana.22640.
[15] Giugno A, Falcone E, Fortunato F, Sammarra I, Procopio R, Gagliardi M et al. Glucose transporter-1 deficiency syndrome with extreme phenotypic variability in a five-generation family carrying a novel SLC2A1 variant. Eur J Neurol 2024; 31: e16325. https://doi.org/10.1111/ene.16325.
[16] Leen WG, Klepper J, Verbeek MM, Leferink M, Hofste T, van Engelen BG et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain 2010; 133: 655-670. https://doi.org/10.1093/brain/awp336.
[17] Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y et al. Molecular findings among patients referred for clinical whole-exome sequencing. Jama 2014; 312: 1870-1879. https://doi.org/10.1001/jama.2014.14601.
[18] Vaudano AE, Olivotto S, Ruggieri A, Gessaroli G, De Giorgis V, Parmeggiani A et al. Brain correlates of spike and wave discharges in GLUT1 deficiency syndrome. Neuroimage Clin 2017; 13: 446-454. https://doi.org/10.1016/j.nicl.2016.12.026.
[19] Leary LD, Wang D, Nordli DR, Jr., Engelstad K, De Vivo DC. Seizure characterization and electroencephalographic features in Glut-1 deficiency syndrome. Epilepsia 2003; 44: 701-707. https://doi.org/10.1046/j.1528-1157.2003.05302.x.
[20] Bourque DK, Cordeiro D, Nimmo GAM, Kobayashi J, Mercimek-Andrews S. Phenotypic and Genotypic Spectrum of Glucose Transporter-1 Deficiency Syndrome. Can J Neurol Sci 2021; 48: 826-830. https://doi.org/10.1017/cjn.2021.3.
[21] Schwantje M, Verhagen LM, van Hasselt PM, Fuchs SA. Glucose transporter type 1 deficiency syndrome and the ketogenic diet. J Inherit Metab Dis 2020; 43: 216-222. https://doi.org/10.1002/jimd.12175.
[22] Tang M, Park SH, De Vivo DC, Monani UR. Therapeutic strategies for glucose transporter 1 deficiency syndrome. Ann Clin Transl Neurol 2019; 6: 1923-1932. https://doi.org/10.1002/acn3.50882.
 Figures
Figures References
References Peer
Peer Information
InformationFigure 1. The DNA sequencing results of the patient. The results suggested a heterozygous point mutation (c.940G>A) in exon 7 of the SLC2A1 gene.
Figure 2. Changes in the patient's blood glucose and blood ketone levels.
Figure 3. Symptoms and the presence of genetic mutation in the family
[1] De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 1991; 325: 703-709. https://doi.org/10.1056/nejm199109053251006.
[2] Anurat K, Khongkhatithum C, Tim-Aroon T, Limwongse C, Thampratankul L. Sleep Disorder: An Overlooked Manifestation of Glucose Transporter Type-1 Deficiency Syndrome. Neuropediatrics 2022; 53: 129-132. https://doi.org/10.1055/s-0041-1736179.
[3] Pawlik W, Okulewicz P, Pawlik J, Krzywińska-Zdeb E. Diagnostic and Clinical Manifestation Differences of Glucose Transporter Type 1 Deficiency Syndrome in a Family with SLC2A1 Gene Mutation. Int J Environ Res Public Health 2022; 19. https://doi.org/10.3390/ijerph19063279.
[4] Klepper J, Akman C, Armeno M, Auvin S, Cervenka M, Cross HJ et al. Glut1 Deficiency Syndrome (Glut1DS): State of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open 2020; 5: 354-365. https://doi.org/10.1002/epi4.12414.
[5] Koch H, Weber YG. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav 2019; 91: 90-93. https://doi.org/10.1016/j.yebeh.2018.06.010.
[6] Wang D, Pascual JM, De Vivo D. Glucose Transporter Type 1 Deficiency Syndrome. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH et al (eds). GeneReviews(®). University of Washington, Seattle Copyright © 1993-2024, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.: Seattle (WA), 1993
[7] Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep 2013; 13: 342. https://doi.org/10.1007/s11910-013-0342-7.
[8] Takahashi S, Matsufuji M, Yonee C, Tsuru H, Sano N, Oguni H. Somatic mosaicism for a SLC2A1 mutation: implications for genetic counseling for GLUT1 deficiency syndrome. Clin Genet 2017; 91: 932-933. https://doi.org/10.1111/cge.12902.
[9] Sapuppo A, Pavone P, Praticò AD, Ruggieri M, Bertino G, Fiumara A. Genotype-phenotype variable correlation in Wilson disease: clinical history of two sisters with the similar genotype. BMC Med Genet 2020; 21: 128. https://doi.org/10.1186/s12881-020-01062-6.
[10] Jiang Q, Xu H, Yan J, Xu Q, Zheng Y, Li C et al. Sex-specific metabolic alterations in the type 1 diabetic brain of mice revealed by an integrated method of metabolomics and mixed-model. Comput Struct Biotechnol J 2020; 18: 2063-2074. https://doi.org/10.1016/j.csbj.2020.07.019.
[11] Wang D, Pascual JM, DC. DV. Glucose Transporter Type 1 Deficiency Syndrome. . Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A editors Seattle, WA: University of Washington, 1993–2019; 2002 Jul 30 [updated 2018 Mar 1] 2018;
[12] Mullen SA, Suls A, De Jonghe P, Berkovic SF, Scheffer IE. Absence epilepsies with widely variable onset are a key feature of familial GLUT1 deficiency. Neurology 2010; 75: 432-440. https://doi.org/10.1212/WNL.0b013e3181eb58b4.
[13] Pong AW, Geary BR, Engelstad KM, Natarajan A, Yang H, De Vivo DC. Glucose transporter type I deficiency syndrome: epilepsy phenotypes and outcomes. Epilepsia 2012; 53: 1503-1510. https://doi.org/10.1111/j.1528-1167.2012.03592.x.
[14] Yang H, Wang D, Engelstad K, Bagay L, Wei Y, Rotstein M et al. Glut1 deficiency syndrome and erythrocyte glucose uptake assay. Ann Neurol 2011; 70: 996-1005. https://doi.org/10.1002/ana.22640.
[15] Giugno A, Falcone E, Fortunato F, Sammarra I, Procopio R, Gagliardi M et al. Glucose transporter-1 deficiency syndrome with extreme phenotypic variability in a five-generation family carrying a novel SLC2A1 variant. Eur J Neurol 2024; 31: e16325. https://doi.org/10.1111/ene.16325.
[16] Leen WG, Klepper J, Verbeek MM, Leferink M, Hofste T, van Engelen BG et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain 2010; 133: 655-670. https://doi.org/10.1093/brain/awp336.
[17] Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y et al. Molecular findings among patients referred for clinical whole-exome sequencing. Jama 2014; 312: 1870-1879. https://doi.org/10.1001/jama.2014.14601.
[18] Vaudano AE, Olivotto S, Ruggieri A, Gessaroli G, De Giorgis V, Parmeggiani A et al. Brain correlates of spike and wave discharges in GLUT1 deficiency syndrome. Neuroimage Clin 2017; 13: 446-454. https://doi.org/10.1016/j.nicl.2016.12.026.
[19] Leary LD, Wang D, Nordli DR, Jr., Engelstad K, De Vivo DC. Seizure characterization and electroencephalographic features in Glut-1 deficiency syndrome. Epilepsia 2003; 44: 701-707. https://doi.org/10.1046/j.1528-1157.2003.05302.x.
[20] Bourque DK, Cordeiro D, Nimmo GAM, Kobayashi J, Mercimek-Andrews S. Phenotypic and Genotypic Spectrum of Glucose Transporter-1 Deficiency Syndrome. Can J Neurol Sci 2021; 48: 826-830. https://doi.org/10.1017/cjn.2021.3.
[21] Schwantje M, Verhagen LM, van Hasselt PM, Fuchs SA. Glucose transporter type 1 deficiency syndrome and the ketogenic diet. J Inherit Metab Dis 2020; 43: 216-222. https://doi.org/10.1002/jimd.12175.
[22] Tang M, Park SH, De Vivo DC, Monani UR. Therapeutic strategies for glucose transporter 1 deficiency syndrome. Ann Clin Transl Neurol 2019; 6: 1923-1932. https://doi.org/10.1002/acn3.50882.
Peer-review Terminology
Identity transparency: Single anonymized
Reviewer interacts with: Editor
Details
This is an open access article under the terms of the Creative Commons Attribution License(http://creativecommons.org/licenses/by/4.0/), which permits use, distribution and reproduction in any medium, provided the original work is properly cited.
Publication History
Received 2025-04-12
Accepted 2025-05-10
Published 2025-05-14